综述:基于进化和物理启发建模的计算蛋白设计
近日,索邦大学、巴黎大学、巴黎城市大学巴斯德研究所、以色列特拉维夫大学联合发表了题为“Computational protein design with evolutionary-based and physics-inspired modeling:current and future synergies“的蛋白设计综述文章。
蛋白质设计的机器学习方法正沿着两条平行轨道快速发展:基于进化的方法和物理启发的方法。两种方法具有高度互补性。

基于进化的方法,前者推断具有所需结构或功能的蛋白质序列集合共享的序列特征。后者使用机器学习surrogates估计关键生化特性,例如结构自由能、构象熵或结合亲和力。
总结
1 基于进化的方法
(1) 目标特性的显著优化需要探索含有来自野生型蛋白的许多突变的序列。然而,据估计多达50%的单点突变对功能有害,导致多个位点突变时成功率呈指数下降。
一种解决方案是将搜索限制在蛋白质自然进化过程中先前遇到或可能遇到的突变或突变组合。例如,Russ等人[9] 利用DCA设计了数百种不同的具有天然功能的脊索酸变位酶,成功率高(~30%)。最近各种机器学习生成模型,在各种酶和纳米设计任务中取得了成功[10,11,12,13,14]。
(2) family-level模型缺点:不能在跨蛋白质家族中得到泛化,因此需要大量序列进行训练。
克服这些限制的一个可能途径是蛋白质语言模型,因为它们可以同时模拟不相关的蛋白质序列集。
2 物理启发的方法
(1)蛋白质设计问题相当于在规定的构象状态下(作为单体结构,与配体结合)找到具有低自由能的序列。基于力场的蛋白质设计的局限性包括计算成本高(基于蒙特卡洛的优化速度慢且效率低)、骨架结构的序列恢复率不令人满意(30%-50%)以及实验成功率有限。
a) Norn等人提出将负对数似然解释为自由能的代理。他们用模型预测的几何特征的概率来近似玻尔兹曼系综中构象的概率。对于具有多个低能构象的序列,trRosetta预测的几何特征分布通常很宽或多峰。因此,trRosetta估计的自由能比Rosetta能量更好地预测折叠到目标构象。
b) 模型预测分布的香农熵可以作为构象玻尔兹曼分布物理熵的代理。
(2) 基于物理的方法的两个基本挑战:1)需要对结构构象空间进行广泛采样以估计热力学量,以及2)探索巨大序列空间的高计算成本。
基于AlphaFold2, AlphaFold2-multimer和RoseTTAfold的幻觉方案其中预测不确定性(pLDDT或PAE)可以用作构象熵的代理。
3 基于进化和物理启发的协同建模
(1)进化模型可以用于快速生成不同的序列库,然后,根据计算密集型物理启发模型中获得的分数对候选序列进行优先排序。
(2) 训练MSA和结构的联合模型。使用进行信息对基于结构的序列生成模型进行微调,或者相反,使用结构信息对进化模型进行正则化。
(3) 将已知结构信息合并为先验,例如使用结构感知transformer模型,如EvoFormer(其中结构作为模板提供)。
(4) 基于ML的分子动力学[60]或神经力场[76]的未来发展可能为理解目标蛋白的高亲和力结合物的合理设计中的挑战,扩展到其他类型的配体提供了重要动力。
01 基于进化的设计
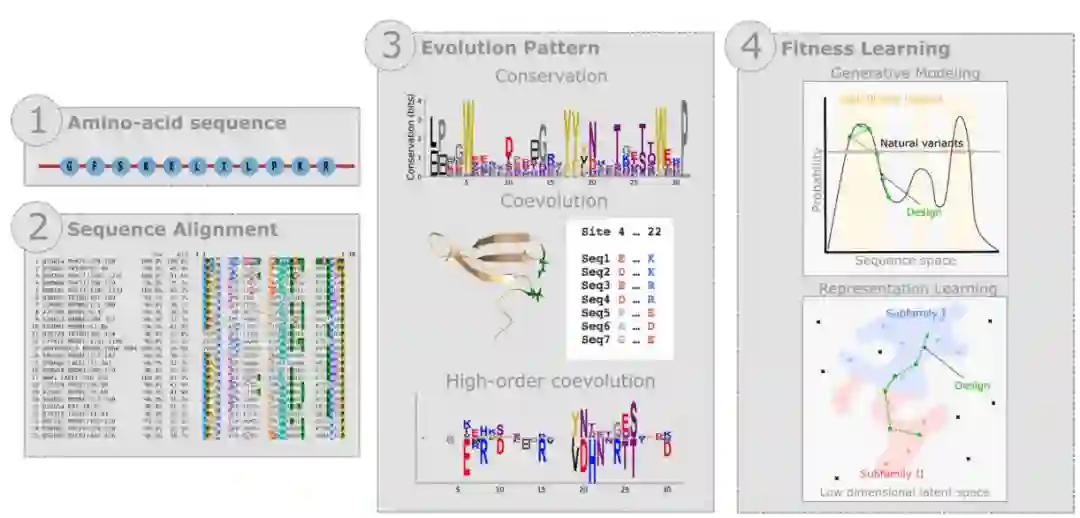
基于进化的设计的主要步骤。从氨基酸序列开始,从序列数据库中检索同源序列,并构建多序列比对(MSA)。MSA具有多种进化模式,包括保守、成对共同进化和反映结构的高阶共同进化以及功能约束。无监督机器学习从进化模式中提取适应度景观fitness landscapes和表示,然后用于设计。
许多设计方案涉及对预先存在的天然蛋白质进行修饰,以获得改进的或新颖的功能特性。目标特性的显著优化需要探索含有来自野生型蛋白的许多突变的序列。然而,据估计,多达50%的单点突变对功能有害,导致多个位点突变时成功率呈指数下降。
一种解决方案是将搜索限制在蛋白质自然进化过程中先前遇到或可能遇到的突变或突变组合。
基于进化的设计包括三个主要步骤:(i)收集和比对一组与野生型蛋白同源的序列,这些序列具有相似的结构和功能,(ii)构建统计/机器学习模型,该模型捕获这些序列之间共享的共同模式,例如保守和共同进化,以及(iii)生成不同于自然序列但保留共同模式的人工序列。
早期的模型,如位置特异性评分矩阵(PSSM)仅专注于捕获位点特异性氨基酸频率,而基于自监督机器学习的越来越复杂的统计模型已经开发出来。
Direct Coupling Analysis(DCA)方法捕获了由协同进化产生的单点和成对相关性,允许对上位效应(epistatic effect)进行建模,并大幅减少搜索空间。
例如,Russ等人[9] 利用DCA设计了数百种不同的具有天然功能的脊索酸变位酶,成功率高(~30%)。基于序列分布的熵,他们估计,在10125个长度相同的可能序列中,1085个是基于单点模型的潜在设计候选,而当包括成对相关性时,只有1025个是合适的。
为了包含额外的高阶统计数据,最近各种机器学习生成模型,在各种酶和纳米设计任务中取得了成功[10,11,12,13,14]。重要的是,这些方法一些还学习到了蛋白质低维潜在空间表示,促进了序列空间的探索和多轮设计小规模实验数据的整合。
除了直接生成新的蛋白质序列,进化模型还可以预测适应度改善突变,指导大规模筛选的文库设计实验,或者相反,从定向进化推断适应度景观实验。
这种family-level模型的一个缺点是它们不能在跨蛋白质家族中得到泛化,因此需要大量序列进行训练。
这对于仅在真核生物中保守的蛋白质尤其成问题。克服这些限制的一个可能途径是蛋白质语言模型,因为它们可以同时模拟不相关的蛋白质序列集。
在大型、未标注的蛋白质序列数据库熵训练的蛋白质语言模型(UniRep, ESM-1b, ProVis, ProtTrans, ProteinBERT)旨在从约10-20%的残基被mask或随机突变的版本重建序列。
蛋白质化学一般理解源于masked语言模型,如氨基酸、二级结构元素或三级接触之间的相似性。然后,可以进一步微调模型,以考虑蛋白质家族的特异性,甚至是具有低序列数和/或多样性的家族。
Hie等人[27] 使用ESM-1b语言模型提出了各种抗病毒抗体的单点突变,并减少了重建误差。在对最佳突变体进行实验表征和重组后,他们发现4/7的测试抗体的结合亲和力可以提高,masked语言模型也可用于MSA,而不是单个序列,如MSA transformer和EvoFormer(AlphaFold的子模块)。
与单序列语言模型相比,MSA级模型显示了改进的序列重建、接触图预测和zero-shot fitness预测,同时需要更少的参数。然而,我们注意到,目前还没有从这些模型生成新序列的既定协议,因为它们不容易定义可直接或通过马尔可夫链蒙特卡洛采样的概率分布。
02 物理启发的方法
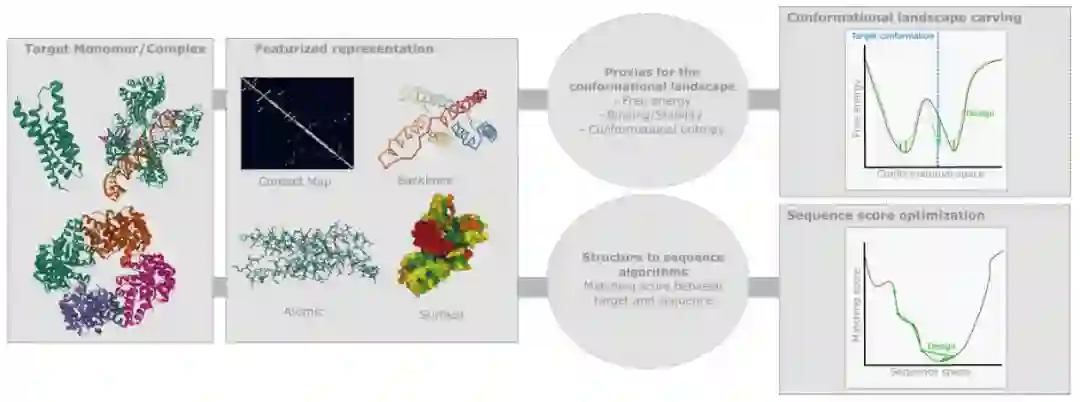
物理启发设计的主要步骤。从(部分或完全规定的)目标结构开始,首先构建适合相关深度学习算法的特征化表示。接下来,两种互补的方法是可能的。首先,可以利用序列到结构预测算法来构建构象景观的代理。后者然后用于设计一个自由能最小值位于目标构象的序列。第二,结构到序列算法可用于从结构生成合适的序列。它们依赖于目标和序列之间的匹配分数,该匹配分数可被优化以找到合适的序列。
蛋白质设计问题相当于在规定的构象状态下(作为单体结构,与配体结合)找到具有低自由能的序列。设计方案的一个常见验证指标是其序列恢复率:从蛋白质的结构开始,剥离其序列定义的侧链,并从剩余的骨架原子重建新序列。序列恢复率是设计序列和原始序列之间的平均序列同一性:高值表明协议很好地概括了结构诱导的序列约束。
为此,已经开发了大量用于蛋白质设计应用的近似力场,包括Rosetta和FoldX。然而,这些力场是启发式,不能faithfully解释潜在的量子动力学和序列从非折叠状态有效折叠到所述结构中能力。
此外,评估自由能还涉及对目标状态之外构象状态的彻底探索。
基于力场的蛋白质设计的局限性包括计算成本高(基于蒙特卡洛的优化速度慢且效率低)、骨架结构的序列恢复率不令人满意(30%-50%)以及实验成功率有限。
在[37]中,Norn等人提出将负对数似然解释为自由能的代理。换句话说,他们用模型预测的几何特征的概率来近似玻尔兹曼系综中构象的概率。尽管模型预测概率相对于构象空间没有精确归一化,但这种近似绕过了对构象空间进行广泛采样以进行自由能估计的要求。他们发现,对于具有多个低能构象的序列,trRosetta预测的几何特征分布通常很宽或多峰。因此,trRosetta估计的自由能比Rosetta能量更好地预测折叠到目标构象。
类似地,模型预测分布的香农熵可以作为构象玻尔兹曼分布物理熵的代理[38]。熵最小化(所谓的幻觉协议)使蛋白质设计具有良好定义的结构,改结构仅部分指定或完全未指定。最近,还提出了基于AlphaFold2、AlphaFold2-multimer和RoseTTAfold的幻觉方案其中预测不确定性(pLDDT或PAE)被用作构象熵的代理[39,40]。这是由于观察到AlphaFold2低置信度预测通常对应于蛋白质的无序区域[41]。
最后,对于蛋白质复合物的设计,Gainza等人[42,43]使用DL计算分子表面pathces之间的匹配分数作为结合亲和力的代理。
重要的是,这些ML模型中的一些模型使得 i)它们的输入蛋白质序列被表示为连续变量(例如,通过one-hot)并且 ii)它们的输出相对于它们的输入是可微的。
因此,可以在一阶近似精度下,在单个反向传播过程中同时评估输入序列的所有单点突变。这使得基于梯度的优化比proposal/rejection Metropolis蒙特卡洛方案更有效[38]。
根据这些方法,一些研究小组成功设计了具有完全指定的或从头骨架结构的蛋白质,并通过双目标部分幻觉围绕功能基序构建折叠。
一个潜在的限制是序列多样性和氨基酸组成偏差。事实上,具有多种构象(例如,多个侧链扭转角)的氨基酸以及因此固有的不确定结构在这种置信度最大化方案中是不利的。因此,这些设计协议可能不包括可以有效采用目标折叠的序列的全部多样性。这些协议的另一个弱点是可能存在“对抗性”最优值:序列对一组网络权重具有高度自信的预测,但对另一组权重没有,即“trick”网络,而不是解决设计问题。
或者,可以尝试直接预测给定折叠的合适序列,即所谓的逆折叠问题。与折叠问题不同,许多序列可以采用完全相同的折叠,因此,应该构造序列的分布。
一种方法是沿着目标骨架"thread“序列,计算序列和backbone之间的兼容性分数,并迭代地对其进行变异以提高其分数。
给定目标骨架构象,Zhou等人[48]基于其组成结构基序及其在蛋白质数据库中的经验氨基酸分布构建了粗粒度统计势。然后,他们通过蒙特卡洛对相应的玻尔兹曼分布进行采样,以生成不同的候选序列。
Anand等人使用三维卷积神经网络,根据当前结构预测可能的氨基酸取代和相应的旋转异构体状态,并迭代突变蛋白质以生成折叠成TIM-barrel。这种协议在数学上等同于从Boltzmann分布采样,其中能量函数是模型计算的负伪似然。
第二种方法是直接建立易于取样的序列分布:基于粗粒度、基于图的蛋白质骨架表示的自回归生成模型允许在单次通过中生成完整序列。这些模型实现了更高的序列恢复率,并且与基于力场的方法相比计算强度低的多。它们已被用于固定骨架单体设计,以及多聚体和抗体设计。
最近,Dauparas等人使用自回归信息传递神经网络成功地设计了各种蛋白质。然而,我们注意到,与目标构象的兼容性并不保证没有其他稳定构象。
对于单体设计,低温取样系统地丢弃不常见的氨基酸排列(例如,溶剂暴露的疏水性氨基酸),可能足以消除替代的稳定构象。然而,尚不清楚这是否足以进行多聚体设计,因为多聚体的设计需要暴露的疏水残基,而疏水残基更容易错折叠。
总得来说,自回归模型与物理模型之间的联系尚不清楚。一方面,它们包括基于物理的先验,如欧几里得变换的局部性和不变性,并且它们学习与物理性质相关的氨基酸之间的相似性,以及有利的缔合(如二硫键、盐桥等)。
进一步表明,leave-one-out条件分布P(s_i | s_-i, backbone)可以预测突变对适应度的影响[39][40],因此可以解释为物理能量的代理。另一方面,序列似然P(s| backbone) 的值取决于序列的解码顺序,不清楚选择哪一个。可以说,自回归重建P(s_1 | backbone)的初始分布更具统计性,而非物理性:该模型根据过去的经验从骨架构型“猜测”氨基酸(例如,蛋白质核心通常由疏水残基组成)。
尽管最近取得了进展,但这些方法仍有一些范围局限:通过构建,它们不适合建模无序蛋白质或片段。变构运动或催化活性的微调仍然是一个主要挑战,因为这些模型是基于静态结构训练的,并且是粗粒度的。
对于这种复杂的功能,仍有望取得进展,特别是基于机器学习的分子动力学[60]的发展,这可以解锁具有复杂动力学行为的蛋白质设计。
另一个令人担忧的来源是这些模型越来越偏离物理学:例如,AllphaFold隐式假设存在分子辅因子、翻译后修饰或蛋白质伴侣,以正确折叠结构。因此,在实验条件下,高度自信的insilico预测可能被证明是错误的,并且模型导出的匹配分数与目标物理特性的识别并不总是正确的。例如,基于trRosetta的幻觉方案[38]设计的一些蛋白质在体外形成同源寡聚体或聚集体,与单体insilico预测不一致。
03 Synergisitc methods
基于进化和物理启发的方法在覆盖范围和范围熵具有高度互补性。虽然受物理启发的模型预测了一般的生物化学性质(单体和蛋白质配体或蛋白质-蛋白质复合物的稳定性),但基于进化的方法以不可知的方式学习了各种特定于家族的功能约束,包括稳定性或催化活性,还包括变构偶联或均低聚物状态的规范。因此,将这两种方法结合起来来以获得最佳成功率是很有吸引力的。这可以通过多种方式实现。
首先,进化模型可以用于快速生成不同的序列库,然后,根据计算密集型物理启发模型中获得的分数对候选序列进行优先排序。示例包括RROSS和FUNCLIB网络服务器,它们使用Rosetta和PSSM信息自动重新设计酶,以提高稳定性或改进催化活性。Tran等人和Das等人分别使用分子动力学的生成模型设计了细胞穿透和抗菌肽。
如果大部分进行设计序列具有令人满意的物理分数,这种简单方法就足够了。否则,可能需要多目标优化/蒙特卡洛采样来生成具有高进化可能性和物理分数的序列。通过优化Rosetta能量和Potts模型估计的进化分数的加权和,重新设计救援协议序列。
一个悬而未决的问题是,是否需要单独的物理和进化模型。相反,我们能否从进化中学习物理相互作用,并反过来从结构预测进化?一个有希望的方向是训练MSA和结构的联合模型,该模型改编自语言和结构预测模型。其他选项可以包括使用进行信息对基于结构的序列生成模型进行微调,或者相反,使用结构信息对进化模型进行正则化。
关于基于进化的模型,需要更多的努力来开发能够在蛋白质家族之间进行推广的模型,同时保持计算可追踪性(对整个社区的可访问性)、可解释性和定义良好的采样协议。
另一个有趣的方向是将已知结构信息合并为先验,例如使用结构感知transformer模型,如EvoFormer(其中结构作为模板提供)。此外,进化模型的训练方案应该更好地考虑样本之间的系统发育关系。Weinstein等人认为,在没有适当处理系统发育的情况下,适应度预测性能可能会随着模型复杂性的增加而稳定或降低,这是最近在语言模型中观察的现象。概念上和实践上的限制是进化约束的纠缠:当前模型不能选择性地丢弃与体内相关但与工程蛋白无关的特定约束(例如,结合抑制剂蛋白的要求,对特定细胞隔室的充分性等)。相反,在家族水平上进化匹配的蛋白质可能不适合特定的生化任务(例如,它们可能结合相关配体,但不同于规定的配体)。
物理启发的方法在设计具有完全或部分特定结构的单体和均低聚物方面取得了令人印象深刻的进展。
尽管取得了显著进展,但目标蛋白的高亲和力结合物的合理设计仍然极具挑战性,总体成功率较低,需要通过定向进化进行后续的体外或体内优化[43,74]。
这源于 i)复合物失败,其中设计的蛋白质可能由于错误折叠、不能结合靶蛋白或不能结合在所需位置而失败 ii)蛋白质-蛋白质相互作用建模中的固有挑战,这涉及部分溶剂化效应,并且经常涉及柔性区域,如免疫球蛋白环或肽。更一般而言,静态构象的粗粒度建模排除了捕获关键功能特征,如无序、中间催化状态或变构运动。
基于ML的分子动力学[60]或神经力场[76]的未来发展可能为更好地理解这些功能开辟道路。扩展到其他类型的配体,如离子、核苷酸或小有机分子也是重要的未来方向。
虽然没有系统地采用基于提议/接受或联合优化的协同方法,但它们证明了成功率的提高,应在短期内更多地考虑。从长远来看,根据结构预测MSA的模型,或者相反,基于结构的进化模型先验,可以克服每种方法的当前限制,例如建模灵活性或进化约束的分离。
参考文献
[9] W. P. Russ et al., An Evolution-Based Model for Designing Chorismate Mutase Enzymes, Science 369, 440 (2020). Evolution-based design. Here, the authors trained a Direct Coupling Analysis generative model on a multiple sequence alignment of chorismate mutase enzymes, and used it to generate a diverse set of artificial sequences. A high fraction (83%) of designed sequences had native-like functionality, despite having limited sequence identity to known natural proteins.
[10] A. J. Riesselman, J. B. Ingraham, and D. S. Marks, Deep Generative Models of Genetic Variation Capture the Effects of Mutations, Nat. Methods 15, 10 (2018).
[11](*)A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Couairon, A. Chen, and D. Bikard, Generating Functional Protein Variants with Variational Autoencoders, PLOS Comput. Biol. 17, e1008736 (2021). Evolutionary-based design. Here, the authors proposed regular and autoregressive variational autoencoders for generative modeling of protein sequences, and tested it for experimental design of luxA bacterial luciferase variants. They found that models trained on aligned rather than unaligned sequences were more successful at generating active sequences. In a second design iteration, they predicted solubility from learnt latent variables and used conditional sampling to generate soluble variants.
[12] J. Tubiana, S. Cocco, and R. Monasson, Learning Protein Constitutive Motifs from Sequence Data, ELife 8, e39397 (2019).
[13] D. Repecka et al., Expanding Functional Protein Sequence Spaces Using Generative Adversarial Networks, Nat. Mach. Intell. 3, 4 (2021).
[14] J.-E. Shin, A. J. Riesselman, A. W. Kollasch, C. McMahon, E. Simon, C. Sander, A. Manglik, A. C. Kruse, and D. S. Marks, Protein Design and Variant Prediction Using Autoregressive Generative Models, Nat. Commun. 12, 1 (2021).
[37] C. Norn et al., Protein Sequence Design by Conformational Landscape Optimization, Proc. Natl. Acad. Sci. 118, e2017228118 (2021).
[38](**) I. Anishchenko et al., De Novo Protein Design by Deep Network Hallucination, Nature 600, 547 (2021). Physics-inspired design In this work, the authors generate a diverse set of sequences with unspecified but stable structures via “network hallucination”, i.e. a conformational entropy minimization scheme based on the trRosetta distogram predictor. About 20% of the designed sequences exhibited a stable fold and of those, the experimentally-determined structure matched well the predicted one. While no novel fold was discovered, hallucination can be used to efficiently scaffold binding motifs.
[39] J. Wang et al., Scaffolding Protein Functional Sites Using Deep Learning, Science 377, 387 (2022).
[40] P. Bryant and A. Elofsson, EvoBind: In Silico Directed Evolution of Peptide Binders with AlphaFold. [41] E. Porta-Pardo, V. Ruiz-Serra, S. Valentini, and A. Valencia, The Structural Coverage of the Human Proteome before and after AlphaFold, PLOS Comput. Biol. 18, e1009818 (2022).
[42] P. Gainza, F. Sverrisson, F. Monti, E. Rodolà, D. Boscaini, M. M. Bronstein, and B. E. Correia, Deciphering Interaction Fingerprints from Protein Molecular Surfaces Using.
[48] J. Zhou, A. E. Panaitiu, and G. Grigoryan, A General-Purpose Protein Design Framework Based on Mining Sequence–Structure Relationships in Known Protein Structures, Proc. Natl. Acad. Sci. 117, 1059 (2020).
[60] X. Fu, T. Xie, N. J. Rebello, B. D. Olsen, and T. Jaakkola, Simulate Time-Integrated Coarse-Grained Molecular Dynamics with Geometric Machine Learning, arXiv:2204.10348.
[76] S.-L. J. Lahey and C. N. Rowley, Simulating Protein–Ligand Binding with Neural Network Potentials, Chem. Sci. 11, 2362 (2020).
专知便捷查看
便捷下载,请关注专知公众号(点击上方蓝色专知关注)
后台回复“EBPM” 就可以获取《综述:基于进化和物理启发建模的计算蛋白设计》专知下载链接