
**——背景——****如何在大尺度的生物分子体系中应用量子化学方法进行准确、快速的计算是长期以来极具挑战性的问题。随着Google Quantum AI近些年来在量子化学模拟上展现的量子优势,基于量子计算机的量子模拟在近些年来已经成为量子化学面向大尺度体系高性能计算最有希望的领域之一。针对这一挑战,QC Ware和勃林格殷格翰(Boehringer-Ingelheim) 在今年一月于Chem. Sci. 联合发表题为"Towards the simulation of large scale protein-ligand interactions on NISQ-era quantum computers"的文章,发展了一套在蛋白-配体复合物体系中计算相互作用能的经典-量子混合方法。作者之一的Robert M. Parrish,是著名的量子化学开源程序Psi4的开发者之一,从佐治亚理工学院David Sherrill组毕业后,在QC Ware公司担任化学模拟研究方向的带头人。——方法——******本文尝试利用量子计算机来突破目前电子结构理论难以广泛应用于大尺度生物分子体系的困境。由此,作者开发了SAPT(VQE)方法,其流程 (如图1所示) 大致可以分为: 1) 预处理步骤: 使用量子算法VQE (variational quantum eigensolver) 获取活性空间 (可简单地看作是配体结合位点) 精确的一阶和二阶约化密度矩阵
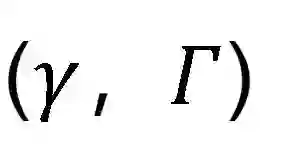
; 2) 后处理步骤: 利用对称匹配微扰理论 (symmetry adapted perturbation theory, SAPT) 计算体系的总能量和各个能量贡献。

**——总结及展望——**本文从第一性原理出发,尝试用量子计算机开展暴力的化学模拟来解决药物设计领域的现实问题—相互作用能计算。即使SAPT(VQE)未展现出明显的量子优势,但是在近期的量子设备上,本文呈现的结果是具启发性的。笔者有如下的简单展望:1. 在低量子比特数的条件下,单层的muCJ在能量成分计算上达到亚kcal/mol精度,意味着SAPT(VQE) 可能可以作为精确的一阶极化能的校正方法;2. SAPT(VQE) 方法可能有利于开发达到量化精度的分子对接打分函数;3. 另外一个值得探索的方向,ansatz的搜索或优化。例如,尝试adaptive ansatz架构,自适应执行高贡献激发,提高量子比特利用率;4. 补充本文不足的工作,引入色散项和诱导项的计算。这一部分工作本文作者已经完成,使用无规相近似来计算两项贡献,感兴趣的读者可阅读参考文献[4]。 参考文献:
1.Malone, F. D.; Parrish R. M.; Welden A. R., et al. Towards the simulation of large scale protein-ligand interactions on NISQ-era quantum computers. Chem. Sci., 13, 3094 (2022). DOI: 10.1039/d1sc05691c. 2.图1来自www.youtube.com/watch?v=8jrb0ZHxdFI 3.Anselmetti, G. L. R., et al. Local, expressive, quantum-number-preserving VQE ansatze for fermionic systems. arXiv:2104.05695. 4.Loipersberger, M., et al. Interaction energies on noisy intermediate-scale quantum computers. arXiv:2207.00218.
点击左下角的"阅读原文"即可查看原文章。
作者: 幻幻 审稿:王凡灏 编辑:林康杰
GoDesign ID:Molecular_Design_Lab ( 扫描下方二维码可以订阅哦!)

本文为GoDesign原创编译,如需转载,请在公众号后台留言。
