冷冻电子显微镜的进展为大分子蛋白质结构测定提供了潜力,但是在多域蛋白质的域间方向建模,成功率仍然很低。近日,Nature子刊,作者使用冷冻电子显微镜开发了自动的域增强建模(DEMO-EM)方法。D****EMO-EM方法通过结合刚体域拟合和柔性装配模拟(具有深度神经网络域间距离分布的灵活装配模拟)的渐进式结构精调程序,从冷冻电子显微镜图中组装多域蛋白结构。

该方法在包含多达 12 个连续和不连续结构域的大规模蛋白质基准集上进行了测试,这些结构域具有中到低分辨率的密度图,其中,对于 97% 的案例,DEMO-EM 生成的模型具有正确的域间方向(模板建模分数(TM 分数)>0.5),并且优于最先进的方法。
DEMO-EM流程图
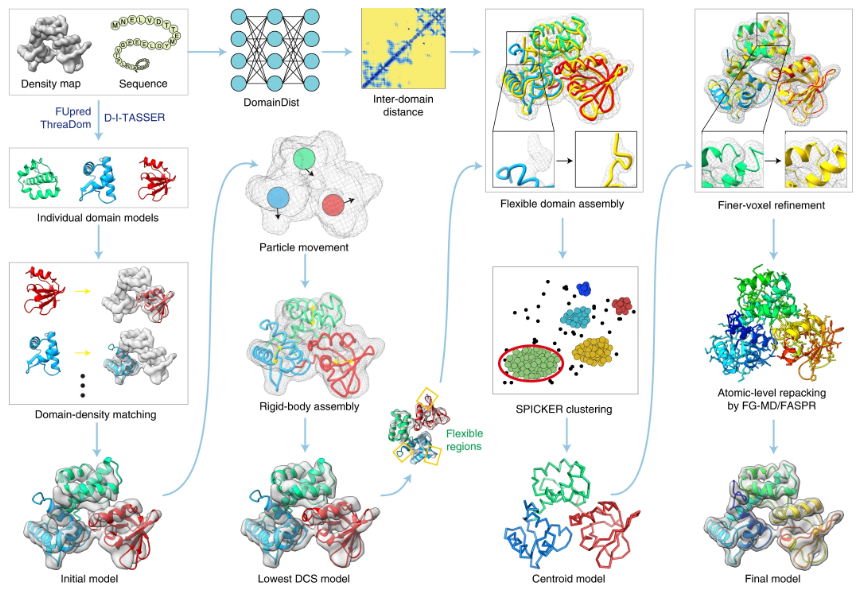
使用结核分枝杆菌依赖性因子的三域蛋白进行说明
从查询序列开始,域边界****首先由FUpred和ThraDom预测,每个域的模型由DI-TASSER生成。同时,使用深度学习卷积网络DomainDist预测域间距离。其次,每个域模型都通过拟牛顿搜索独立地拟合到密度图。第三,初始全长模型通过两步刚体REMC模拟优化****,以最小密度图和全长模型之间的DCS。第四,使用由DCS、域间距离分布和基于知识的力场引导的REMC模拟,通过原子级、分段级和域级精调的柔性装配对从刚体装配模拟中选择的最低DCS模型进行精调,得到decopy构象由SPICKER聚类以获得质心模型。最后,对全原子模型再次进行柔性装配模拟,其中质心模型的约束增加了能量,最终模型是用FASPR和FG-MD侧链重新包装后的最低能量模型构建。
DEMO-EM是一种基于冷冻电镜图的多域蛋白质结构确定的分层方法,由四个连续步骤组成:(1)确定域边界并对单个域建模,(2)将域模型与密度图匹配初始框架生成,(3)用于域位置和方向优化的刚体域结构组装和(4)全长结构模型的柔性结构组装和细化模拟。
从冷冻电镜合成密度图构建多结构域蛋白
从查询的氨基酸序列开始,首先应用LOMETS**[1]从PDB创建多个模版比对,其中ThreaDom用于根据域保守分数预测域边界。如果蛋白质被LOMETS定义为“简单”目标,并且比对覆盖率>95%,则应用ThraDom预测的域定义。否则,通过FUpred(通过最大化域内接触的数量[3]和最小化由基于深度学习的神经网络ResPRE[2])**预测的接触图上的域间接触的数量来预测域边界。
接下来使用DI-TASSER**[4]生成每个域的结构模型,它是I-TASSER[5]**的一个版本,通过将深度学习预测的残基间接触和距离图以及氢键电位结合到迭代搜索全基因组和宏基因组序列数据库来构建多序列比对(MSA)。
然后根据TripletRes预测的接触选择最前面的MSA,将其输入到ResPRE**[2],**TripletRes是基于深度残基神经网络预测距离图、氢键网络和扭转角。这些预测的约束被集成到I-TASSER力场中以指导replica-exchange 蒙特卡洛模拟(REMC)。
最终模型由SPICKER聚类并由FG-MD改进。对于包含来自查询序列不同区域的两个或多个片段的不连续域,域模型是通过顺序连接所有片段的序列获得的。
基于深度神经网络的域间距离预测
为了帮助指导域方向组装,域间距离图由深度残差神经网络算法 DomainDist 预测,DomainDist 是TripletRes 的扩展,最初开发它是为了基于共进化矩阵的三元组预测残基间接触图,但在这里扩展以预测 2-20 Å 范围内 36 个 bin 内残基间距离的概率。
DomainDist 程序在从 PDB 收集的 26,151 种蛋白质的非冗余数据集上进行训练,其中每种蛋白质的 MSA 是使用 HHblits搜索 Unilust30 序列数据库构建的. 除了 TripletRes 中采用的二维 (2D) 协同进化特征外,还采用了三个一维 (1D) 特征,包括隐马尔可夫模型、Potts 模型的序列和场参数的 one-hot 表示并平铺到两个维度并与二维协同进化特征连接。
神经网络结构是按照卷积策略设计的,使用 ResNet 基础模块。神经网络模型由 Adam 优化算法训练,以最小化交叉熵损失。尽管 DEMO-EM 只考虑了域间距离信息,但在训练过程中同时考虑了域内和域间距离信息。
基于拟牛顿的域匹配和冷冻电子密度图
对于来自 DI-TASSER 的每个单独的域模型,我们使用有限内存 Broyden-Fletcher-Goldfarb-Shanno (L-BFGS),一种具有六维 (6D) 平移-旋转自由度的准牛顿优化算法,来识别与密度图相关性最高的域的最佳位置和方向。
由于L-BFGS是一种局部优化方法,其结果取决于初始解,因此作者通过枚举欧拉角的所有组合( φ, θ and ψ ),以步长Srot_ang穿过密度图空间。
对于domain pose,密度相关分数(density correlation score,DCS)
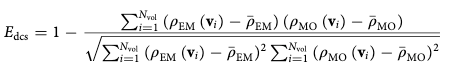
用于指导L-BFGS模拟。Nvol是voxels的个数(网格点),ρEM (vi ) 是第i 个voxel的实验密度。decopy结构探针密度定义为:
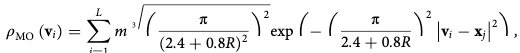
刚体域组装
执行两轮刚体域组装模拟以优化域位置和方向。在第一轮中,这些域被视为粒子,并进行快速的REMC模拟,以根据全局模拟密度相关性调整各个域的位置。第二轮刚体REMC模拟用于微调域位姿,其具有更详细的能量立场。
原子级灵活的域组装和细化
灵活的域组装和细化过程包含两个阶段的模拟,具有渐进的voxels分辨率和采样焦点。在第一阶段,实施了6种不同的动作,(1)LMProt 扰动,(2)围绕连接两个末端的轴的片段旋转,(3)片段沿序列的构象移位,(5)刚体段平移,(5)刚体尾部旋转和(6)刚体域级平移和旋转。
第二阶段,使用在所有原子上计算的DCS实现Voxel大小为2 Å 的更精细的密度图。此外,所有残基都有相同的概率被选中进行移动和采样。当交换次数达到 100 时,模拟终止。选择最低能量的诱饵来构建最终模型,侧链原子由 FASPR 重新包装,然后是 FG-MD 细化。
结果
表2显示了从同一组预测域模型开始时从 MDFF 和 Rosetta 获得的结果,其中初始构象由 Situs 和 MAINMAST 建模组装。这些数据再次表明,DEMO-EM 的表现优于 MDFF、Rosetta 和 MAINMAST,全长模型的平均 TM 分数分别比 MDFF、Rosetta 和 MAINMST 高 60.0%、87.2% 和 144.4%。
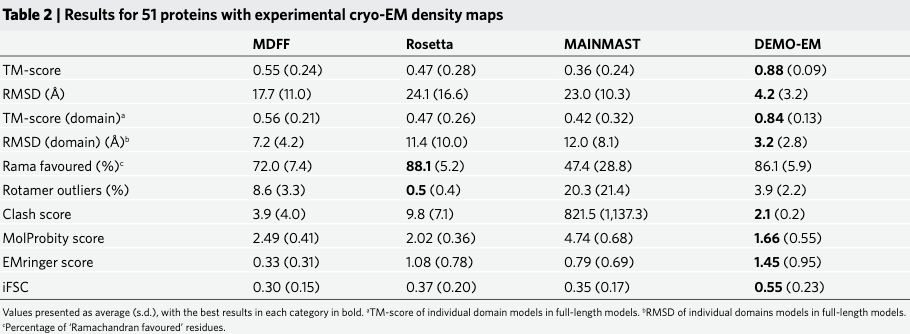
最后,作者在所有 51 个案例上将 DEMO-EM 与最先进的端到端深度学习结构预测方法 AlphaFold2 进行了比较。
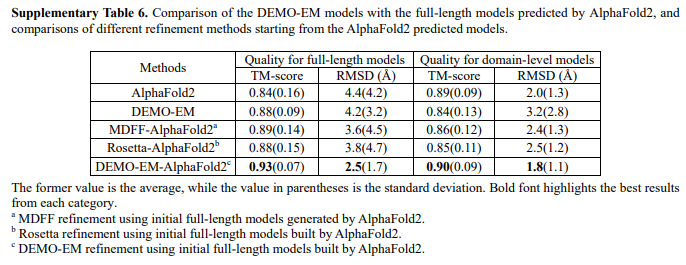
由上表所示, 虽然 AlphaFold2 预测的单个域的 TM-score (0.89) 比 DEMO-EM (0.84) 高,这可能是因为 DI-TASSER 构建的域模型质量较低,整体质量DEMO-EM 构建的全长模型(TM-score 为 0.88)优于 AlphaFold2(TM-score 为 0.84),DEMO-EM 在 28 出时获得了比 AlphaFold2 更高的 TM-score 51 种蛋白质。
作者还将 AlphaFold2 构建的相同全长模型输入 MDFF、Rosetta 和 DEMO-EM,以检查灵活组装和细化过程的性能。所有方法都改进了初始全长模型,即使对于最佳预测模型,也显示了冷冻电镜数据的有用性。
源代码
https://zhanggroup.org/DEMO-EM/
参考资料
[1] Zheng, W. et al. LOMETS2: improved meta-threading server for fold-recognition and structure-based function annotation for distant-homology proteins. Nucleic Acids Res. 47, W429–W436 (2019) [2]Li, Y., Hu, J., Zhang, C., Yu, D.-J. & Zhang, Y. ResPRE: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics 35, 4647–4655 (2019) [3] Zheng, W. et al. FUpred: detecting protein domains through deep-learning based contact map prediction. Bioinformatics 36, 3749–3757 (2020) [4] Zheng, W. et al. Protein structure prediction using deep learning distance and hydrogen‐bonding restraints in CASP14. Proteins 89, 1734–1751 (2021) [5]Yang, J. et al. The I-TASSER suite: protein structure and function prediction. Nat. Methods 12, 7–8 (2015).