传统的计算生物学方法是利用近似的经典势能来计算分子的性质。原子间的相互作用是由物理原理或与实验数据拟合的能量函数来描述的。它们的函数形式通常仅限于原子之间的成对相互作用,而不考虑复杂的多体效应。最近,神经网络已经成为描述生物分子之间相互作用的另一种方式。在这种方法中,能量函数没有显式的函数形式,而是从原子或量子水平的模拟中自底而上学习的。上海理工大学上海高级免疫化学研究所的Francesco Zonta和杨欢等人于最近在JCTC上发表了名为Construction of a Deep Neural Network Energy Function for Protein Physics一文,他们尝试了一种自上而下的方法,并使用深度学习方法,利用在结构生物学领域多年来获得的大量实验数据来获得能量函数,并将其命名为NNEF(Neural network energy function)。
—****—背景介绍——
对大型生物分子的精确计算建模应该涉及到量子力学(QM),但它的计算成本限制了这种方法的可行应用。基于近似经典势能的模型在描述生物大分子的动力学模型及如何相互作用方面更为成功。目前出现了两种主流的简化势能:基于物理模型的势能函数和基于知识的势能函数。这两种方法都依赖于物理直觉,将原子之间的相互作用映射为依赖于一组参数的简单函数形式,并通过QM和实验数据进行参数化。当然,对于一些特殊的体系,简单函数形式不能准确描述系统所需的所有细节。并且,基于经典势能函数的模拟对于较大的生物学体系,在计算上过于昂贵,不能总是达到与实验直接比较的时间和大小尺度,QM计算和混合方法(QM/MM)更是如此。 近年来,出现了将深度学习与基于物理的方法相结合的新方法。例如,在材料化学中,神经网络和量子蒙特卡罗方法已被应用于求解薛定谔方程,或通过拟合基于密度泛函理论或从头算分子动力学的计算来产生经典的原子力场。在蛋白质科学中,一些研究小组已经开发了用于蛋白质多尺度建模的神经网络能量函数,采用自下而上的方法,例如,将粗粒度势拟合到原子MD模拟中。尽管这些能量函数不能转移到不同的蛋白质系统中,但它们可能会导致未来力场参数化方式的范式转变。在本研究中,作者提出了一种自上而下的方法,并使用无监督的深度学习从实验数据(蛋白质结构和序列)中构建一个统计势。统计势并不是一个简单的函数形式,而是由一个深度神经网络来表示。 ——方法——
NNEF算法总览:
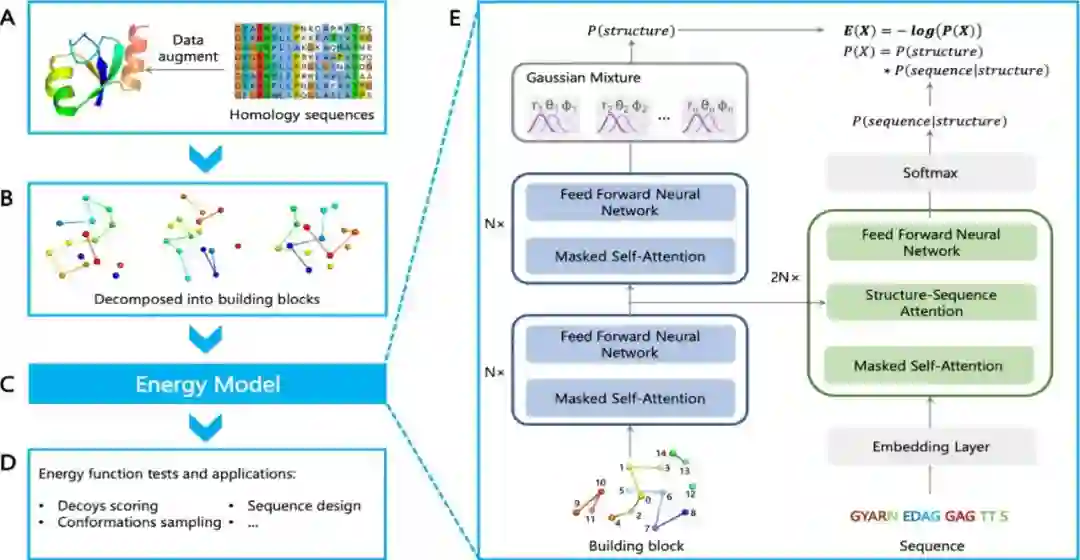
图1 NNEF的流程图 作者首先构建了一个非冗余蛋白质结构数据库,并用它们的同源序列增加训练数据。然后,作者根据每个氨基酸残基周围的局部结构定义了结构模块,用作能量模型的输入。每个结构模块包括残基本身,它沿着序列上的四个最近的残基,以及3D空间中的其他10个最近的残基,每个残基都用一个节点表示,作为训练网络的样本的构建块数量约为25亿个。能量模型如(E)所示,其中作者拟合了一个概率密度函数,并计算出能量为E(X)=−logP(X)。蛋白质的总能量是所有砌块的能量之和。在自回归模型中,作者分离了结构特征和序列特征,并使用Transformer网络架构计算下式:
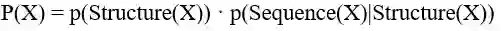
作者使用softmax函数来计算离散特征的概率,并使用高斯混合模型来计算连续特征的概率,学习到的能量函数可以应用于各种下游任务,如decoy评分、构象采样和序列设计。数据集的收集:
从PISCES CulledPDB数据库中采样,序列的identity <= 50%,分辨率<=3.0Å,r因子<=1.0,得到链的总数约为29000。过滤出x射线晶体学解决的结构并将所有的链匹配到HH-suite PDB70数据库中,得到对齐后的链的数量约为19,000条。作者使用hh-filter过滤对齐的序列,要求对PDB序列identity <50% 和 >50% 的coverage。过滤后,作者通过简单地将对齐的序列映射到结构的坐标来生成同源结构(数据集包含全α,全β和α/β结构)。 算法补充说明如图一中所示的算法框架中,这里做几点补充说明:
- 结构单元的选取:
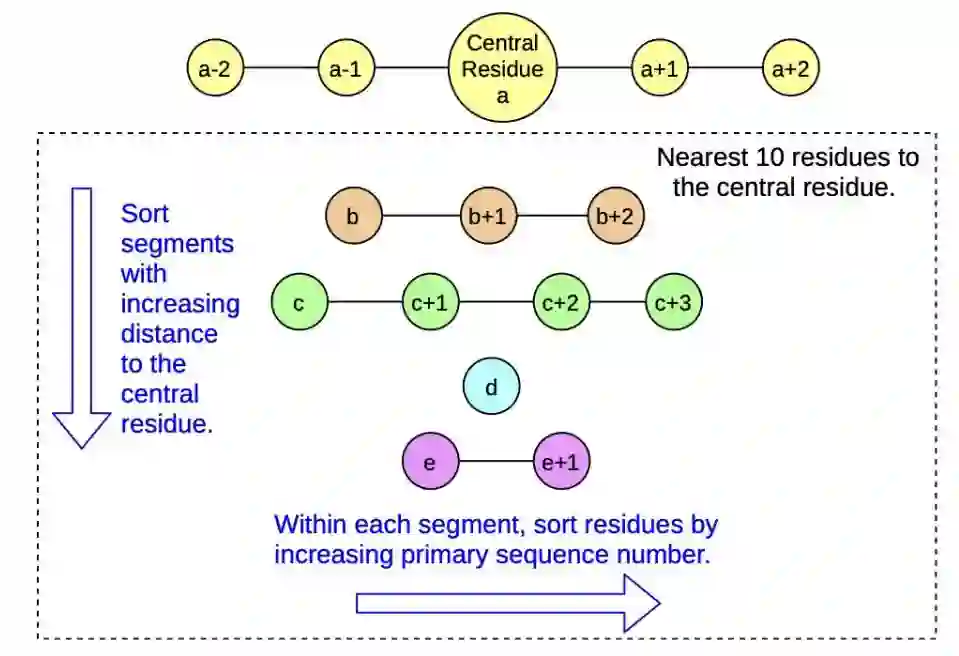
图2 结构基元选取示意图首先从一个残基出发,取最近的几个氨基酸残基作为起始残基,对于起始残基,会沿链的方向取相邻的残基,总数为十个,如图二所示。 2. 对于网络输出的不连续的物理量,模型会通过一个高斯核模型来统一描述:

c,μ,σ分别为该物理量的系数,均值和平方。 ——结果与分析——
首先作者在3DRobot数据库中找出了二级结构为α-helix,β-sheet,α/β的200个结构,每个蛋白取出300个decoy进行打分:
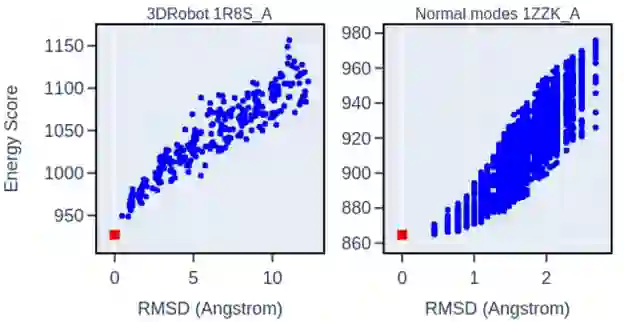
图3 NNEF打分能力评估算例 左:3DRobot诱饵集中一个典型的蛋白质打分的例子。右:简正模decoy集中一个典型的蛋白质打分的例子。红方块是native的结构,蓝点是decoy。在两个诱饵组的所有蛋白质中,可以看到能量随着与native构象与decoy的RMSD的增加而增加。 其次,作者利用NNEF充当蛋白质结构预测的打分依据: 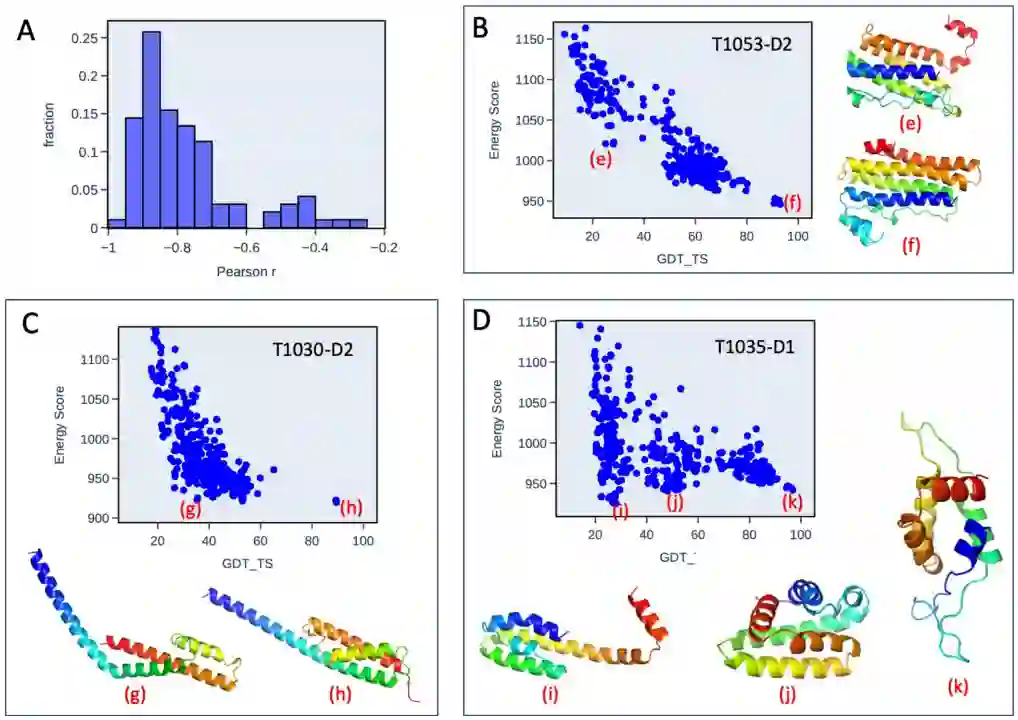
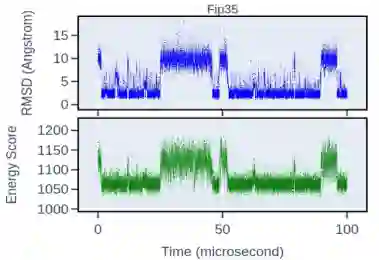
图5 Fip35的MD轨迹能量与RMSD示意图 作者根据模型的输出数据进行Langevin动力学模拟,按照下式更新蛋白质构象: 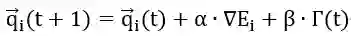
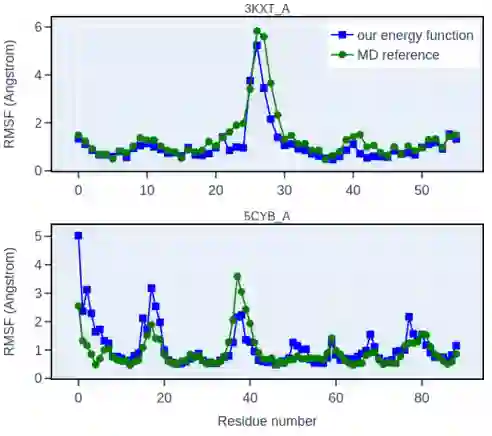
图6 MD模拟结果对比图 朗之万动力学模拟的RMSF。作者将来自NNEF(蓝色)的轨迹的RMSF与用amber-14SB力场(绿色)获得的经典MD模拟的轨迹的RMSF进行了比较。展示了两个高度相关的例子。对于样本中的大多数蛋白质,在整个序列中均能得到具有良好相关性的RMSF。最后作者以NNEF为打分函数,测试了其在序列设计方面的性能,主要是通过在退火过程的每一步,引入一个蛋白质序列的随机点突变,根据Metropolis算法来接受或拒绝该突变,而达到序列设计的目的:
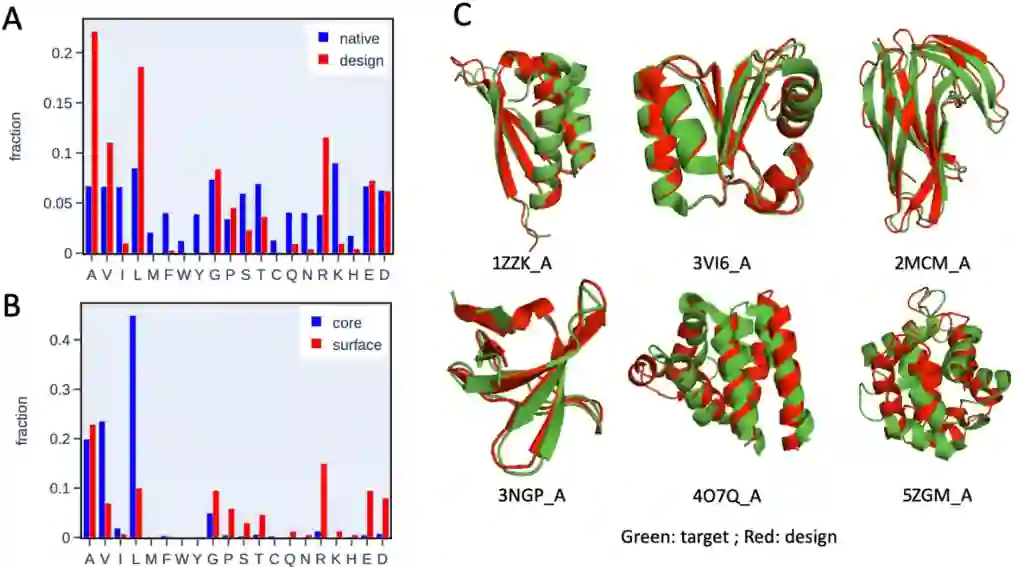
图7 序列设计测试图 (A):1800个设计序列和18个蛋白质的测试样本的天然序列(每个蛋白质100个序列)的氨基酸丰度直方图。设计的序列显示出对10种氨基酸(Ala、Val、Leu、Gly、Pro、Ser、Thr、Arg、Glu和Asp)的偏好性。(B):在所设计蛋白的核心和表面的残基的氨基酸频率。核心残基多为疏水基(Ala、Val和Leu),而极性残基主要分布在表面。(C):使用TrRosetta预测的目标结构和预测结构的几个例子。对于大约三分之二的设计序列,预测的结构可以匹配目标结构。 **——总结——**在本工作中,作者采取一种top-down的方式,使用无监督深度学习方法来推导描述蛋白质内氨基酸相互作用的统计势能,将蛋白质表示为包含三维空间中附近残基的组成块的集合,广泛地探索了每种构建块的构象空间。 目前类似的工作也是有的,但是模拟速度仍然受限,并且文章中对于模型怎么将不同的模块串联而进行总体能量更新的方式并没有详细的描述,训练方式与源代码也没有进行公布,这也是笔者对于该模型存在的一点疑问。 参考文献Huan Yang, Zhaoping Xiong, and Francesco Zonta. Journal of Chemical Theory and Computation[J]. 2022. Article ASAP DOI: 10.1021/acs.jctc.2c00069
点击左下角的"阅读原文"即可查看原文章。
作者:王凡灏审稿:幻 幻编辑:王丽莹 GoDesignID:Molecular_Design_Lab( 扫描下方二维码可以订阅哦!)