
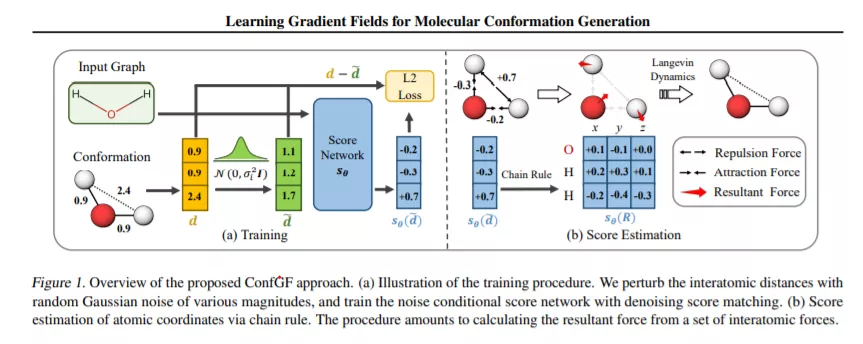
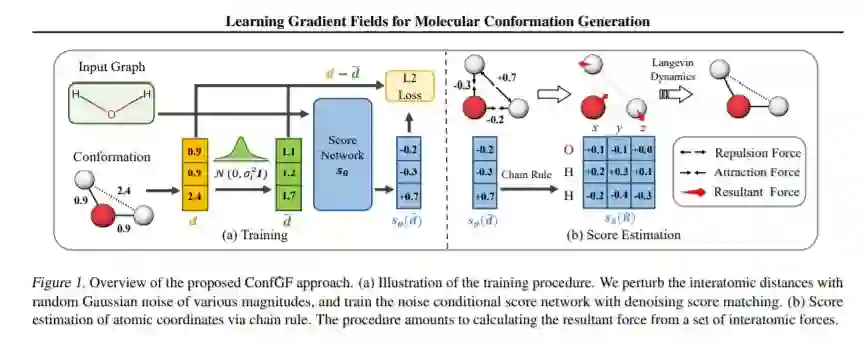
我们研究计算化学中的一个基本问题,即分子构象生成,试图从二维分子图中预测稳定的三维结构。现有的机器学习方法通常首先预测原子之间的距离,然后生成满足这些距离的3D结构,而在3D坐标生成过程中,预测距离中的噪声可能会导致额外的误差。本文受传统分子动力学力场模拟方法的启发,提出了一种直接估算原子坐标对数密度梯度场的新方法ConfGF。估计的梯度场允许通过朗之万动力学直接生成稳定的构象。然而,由于梯度场是旋转平移等变的,因此该问题非常具有挑战性。我们注意到估计原子坐标的梯度场可以转化为估计原子间距离的梯度场,因此开发了一种基于最近的基于分数的生成模型的新算法来有效地估计这些梯度。跨多个任务的实验结果表明,ConfGF显著优于以前的最先进基线。
https://arxiv.org/abs/2105.03902
成为VIP会员查看完整内容
相关内容

相关资讯
相关论文