
准确预测分子性质有助于评估和选择具有许多下游应用所需特性的合适化学分子。随着近年来图神经网络(GNNs)在各种图相关任务中的显著成功,已经从不同方向进行了许多努力来设计用于分子性质预测的GNN模型。基本思想是将原子和键的拓扑结构视为一个图,并使用强大的GNN编码器将每个分子转换为一个表示向量,然后设置特定属性的预测模块。
https://www.zhuanzhi.ai/paper/79ef70c82681e1e2ed1c394d8550bf47
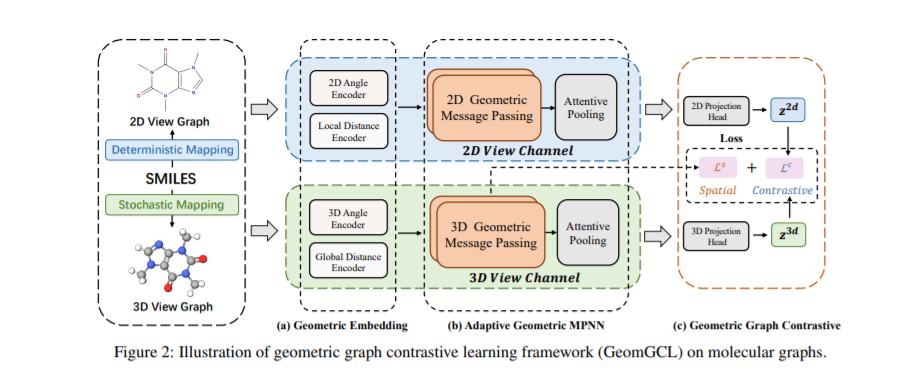
当缺乏足够的标记数据时,图对比学习(GCL)方法在许多应用中显示出非常好的性能。标记数据的缺乏是用于分子性质预测的GNN模型(以及其他深度学习模型)预测性能的主要障碍之一。现有的GCL方法通常对图采用不同的数据增强方案,当其应用到其他领域的图上时可能会改变图的语义。目前大多数关于分子图的GCL方法仍然基于这样的数据增强方法,这不可避免地会改变分子的天然结构。例如,(You 等人)提出丢弃原子、扰动边缘和屏蔽属性来增强数据。然而,由于每个原子都对分子性质有影响,原子的这种随机删除和扰动会破坏分子的结构。虽然像MoCL这样的一些其他方法采用预定义的分子子结构来缓解随机破坏的问题,但这种替代规则仍然有可能违反化学原理。
本文提出了一种用于分子性质预测的几何增强的图对比学习模型(GeomGCL),该模型配备了自适应几何消息传递网络(GeomMPNN)以及增强2D-3D几何结构学习过程的对比学习策略。该框架可以在不破坏分子结构的情况下,从不同的几何角度预测分子性质。
成为VIP会员查看完整内容
相关内容
Arxiv
10+阅读 · 2021年10月4日

相关VIP内容
相关资讯