
编译 | 陈睿哲 本文介绍一篇拜罗伊特大学2022年7月发表在nature communications的《ProtGPT2 is a deep unsupervised language model for protein design》。蛋白质设计在自然环境和生物医学中发挥着重要作用,旨在为特定用途设计全新的蛋白质。受到近期Transformer架构在文本生成领域成功的启发,作者提出ProtGPT2,一种在蛋白质空间上训练的语言模型,用于生成遵循自然序列原则的全新蛋白质序列。ProtGPT2生成的蛋白质显示出天然氨基酸倾向,而无序预测表明,88%的ProtGPT2生成的蛋白质是球状的,与自然序列一致。蛋白质数据库中的敏感序列搜索表明,ProtGPT2序列与自然序列有着远亲关系,相似网络进一步证明,ProtGPT2是对蛋白质空间中未探索区域的采样。ProtGPT2生成的序列在探索蛋白质空间的未知区域时,保留了天然蛋白质的关键特征。

1 简介 近年来,预训练大模型极大地推动了自然语言处理领域的发展。作者等人注意到,蛋白质序列与人类语言有着某种相似性。蛋白质序列可以描述为化学定义的字母、天然氨基酸的串联,与人类语言一样,这些字母排列形成二级结构元素(“单词”),单词的集合形成承担功能的“句子”。蛋白质序列与自然语言一样,是信息完整的:它们以极其高效的方式完全按照氨基酸顺序存储结构和功能。随着自然语言处理领域在理解和生成具有接近人类能力的语言方面的非凡进步,作者假设这些方法为从序列的角度处理蛋白质相关问题打开了一扇新的大门,例如蛋白质设计。
受到自回归语言模型(如GPT系列)和先前采用自回归语言模型建模蛋白质序列的成功,作者想知道能否通过自回归语言模型来建模蛋白质序列,以达到:1)高效地学习蛋白质序列语言;2)生成合适且稳定的蛋白质;3)理解所生成的序列与自然语言处理的关系,模型能否对未知蛋白质空间进行采样。
因此,作者提出了ProtGPT2,一种具有7.38亿参数的GPT架构的自回归模型,能够以高通量方式生成从头蛋白质序列。ProtGPT2在整个蛋白质空间的百万序列上进行训练后,有效地学习了蛋白质语言。ProtGPT2生成的蛋白质序列具有与自然序列相同的氨基酸和无序倾向,同时在进化上远离当前的蛋白质空间。二级结构预测计算出88%的序列是球状的,与天然蛋白质一致。使用相似网络表示蛋白质空间表明,ProtGPT2序列通过扩展自然超家族来探索蛋白质空间的未知区域。生成的序列显示出与自然序列类似的预测稳定性和动态特性。由于蛋白质设计在解决从生物医学到环境科学等领域的问题方面具有巨大潜力,作者认为ProtGPT2是高效高通量蛋白质工程和设计的重大进步。
2 方法 分词与词表 作者通过BPE分词器在数据集上训练,最终的词表大小为50256个tokens,每个token平均包含了4个氨基酸。
数据集 作者以Uniref50作为训练数据集,包含49874565个序列。随机选择10%的序列生成验证数据集。最终的训练和验证数据集分别包含4488万和499万个序列。作者生成了两个数据集,一个使用512的序列大小,另一个使用1024。本文中展示的结果对应于使用512大小训练的模型(后文主模型)。
模型 作者使用堆叠Transformer decoder作为模型架构,ProtGPT2模型由36层Transformer decoder组成,模型维度为1280。在自回归语言模型的条件下,每个句子的概率分布被定义为如下:
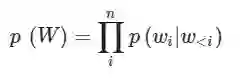
更具体的,作者通过优化句子中每个单词与其前缀的负对数似然作为目标函数来进行优化:
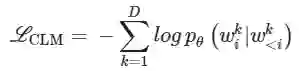
模型训练 输入序列为BPE分词器所分词后得到的token序列。模型权重在训练之前重新初始化。使用Adam(β1)优化模型 = 0.9, β2 = 0.999),学习率为1e-03。对于主模型,每个batch为512 tokens × 128 GPUs。每个GPU处理8个batch,总计1024个。模型在128个NVIDIA A100上训练4天。使用DeepSpeed69进行并行训练。
模型推理 作者使用主模型,使用不同的推理参数,对序列进行采样。在采样策略与采样超参数上,重复惩罚在1.1到3.0之间以0.1进行微调,选择token的范围top-k从250到1000不等,每50采样一次,并且top-p从0.7到1.0,窗口为0.05个单位(top-k采样,模型会从概率前 k 大的单词中抽样选取下一个单词;top-p采样,设定概率阈值,取满足阈值条件的样本进行采样)。基于以上的参数设定,为每种采样参数生成100个序列,并将其氨基酸的频率与自然序列进行比较。作者观察了哪些参数在自然序列中七种最常见的氨基酸组中产生的差异较小。作者还探索了50到100范围的beam search采样,但在所有情况下都会产生较差的结果。为了确定自然序列中的氨基酸频率以与ProtGPT2样本进行比较,作者从Uniref50数据集中随机选取了100万个序列。作者通过微调超参数找到了最佳匹配参数,某个采样过程如图1所示。
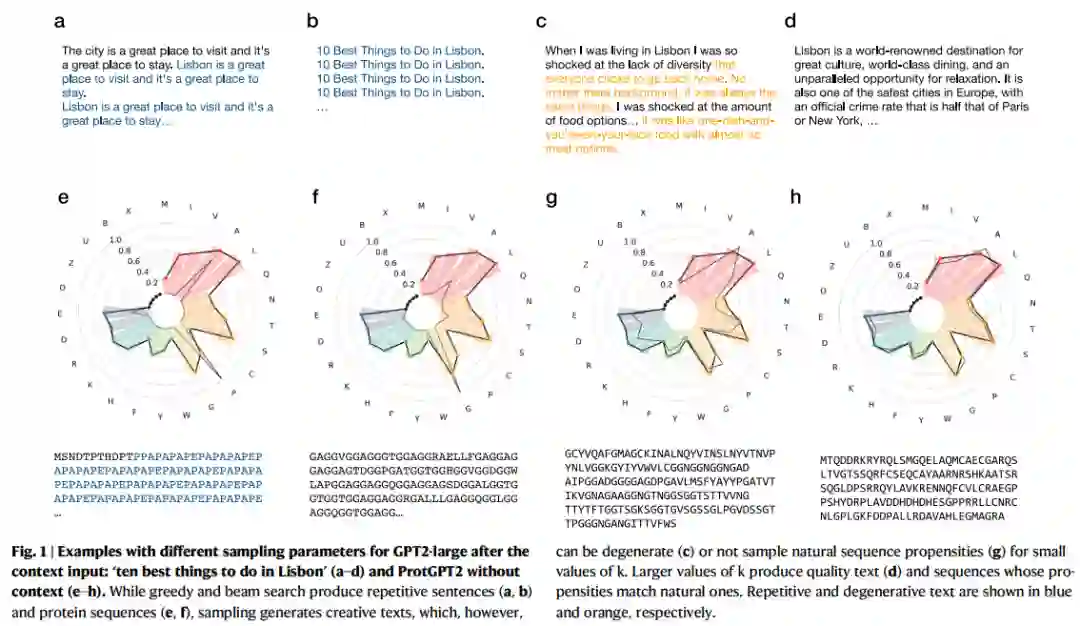
3 结果 统计采样 自回归语言生成基于以下假设:序列的概率分布可以分解为条件下一个词分布的乘积。然而,一个训练好的语言模型生成不连贯的乱码或重复文本并不罕见。作者简要总结了本研究中应用的最常用的语言生成采样策略与超参数。
贪婪搜索策略是在每次采样时选择概率最高的token。虽然算法简单,但生成的序列是确定性的,很快也会变得重复(图1a)。Beam search试图通过保留最可能的候选词来缓解这一问题,尽管生成的文本仍然存在重复性,人类文本往往会交替使用低概率和高概率token(图1b)。最后,通过从前k个最可能的词中随机选取一个词,随机采样远离确定性采样(图1c,d)。
根据先前关于语言模型采样策略的研究,受这项工作的启发,作者按照不同的采样策略和采样超参数生成序列(图1)。为了评估什么样的采样过程产生了最自然的相似序列,作者将生成的序列集的氨基酸倾向性与在自然蛋白质序列中发现的进行了比较。作者还观察到贪婪搜索和Beam search会产生重复的确定性序列,而随机采样显著改善了生成的倾向性(图1)。此外,作者还观察到,生成类似于自然序列的序列需要较高的k值,即最佳结果出现在k > 800的范围内,作者特别选择了k = 950(图1h)。作者还发现,当选择1.2的重复惩罚时,采样结果得到了改善。因此,本研究的其余部分使用了这些采样参数。
ProtGPT2序列编码球状蛋白 为了在序列和结构属性的背景下评估ProtGPT2生成的序列,作者创建了两个数据集,一个是使用前面描述的推理参数从ProtGPT2生成的序列;另一个是从UR50随机选择的序列。每个数据集由10000个序列组成。由于ProtGPT2是以无监督的方式训练的,作者的分析重点是验证ProtGPT2序列的结构和生化特性。
作者首先研究了数据集中的无序和二级结构内容。之前已经表明,在细菌和古细菌中发现的蛋白质中约有14%是无序的。为此,作者运行IUPred335来分析ProtGPT2生成的序列是否比一组自然序列更容易无序。作者的分析显示,在ProtGPT2生成的序列(87.59%)和自然序列(88.40%)中,球状结构域的数量相似。已经报道了几种检测短内在无序区域的方法。由于作者的目标是提供跨数据集的球状度和普遍无序的高水平比较,作者进一步使用IUPred3在氨基酸水平上分析了蛋白质序列。值得注意的是,作者的结果显示两个数据集的有序/无序区域分布相似,ProtGPT2和自然数据集中分别有79.71%和82.59%的有序氨基酸(表1)。
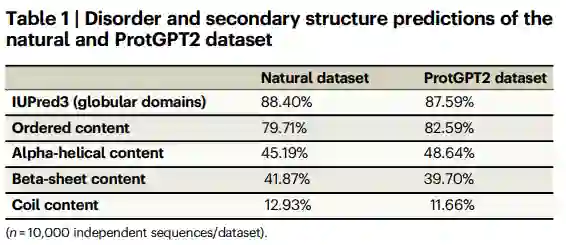
接下来,作者研究了无序中的相似性是否是等效二级结构元素含量的结果。为此,作者计算了ProtGPT2和自然序列数据集的PSIPRED预测。自然序列显示α螺旋、β片和线圈含量分别为45.19%、41.87%和12.93%。ProtGPT2数据集的百分比分别为48.64%、39.70%和11.66%。这些结果表明,ProtGPT2生成的序列类似于球状结构,其二级结构与在自然界中发现的类似。
ProtGPT2序列与自然序列 蛋白质在进化过程中通过点突变以及复制和重组发生了巨大变化。然而,通过序列比较,即使两种蛋白质的序列有显著差异,也可以检测出它们之间的相似性。作者想知道ProtGPT2序列与自然序列的关系如何。为此,作者使用了HHblits,这是一种敏感的远程同源性检测工具,使用配置文件隐马尔可夫模型根据数据库搜索查询序列。作者根据Uniclust30数据库搜索ProtGPT2数据集中10000个序列的同源性。为了进行比较,作者还使用相同的设置对自然数据集执行了相同的搜索。此外,为了分析完全随机序列与ProtGPT2序列的比较情况,作者还通过随机选取词表中的25个字母进行连接,构建了第三个数据集。
因为作者想对数据集与现代蛋白质空间的相关性进行定量比较,所以作者绘制了个体与序列长度图(图2)。具体来说,对于Uniclust30中发现的每一条路线,作者描述了具有最高个体和序列长度的路线。作为序列同一长度空间中的参考点,作者使用HSSP曲线,一个边界集来定义蛋白质序列相关性的置信度。其特性低于该曲线的蛋白质,不一定具有类似的3D结构,也不可能具有同源性。由于ProtGPT2和随机数据集中的序列不是蛋白质进化的结果,作者使用曲线作为已知阈值来比较数据集。
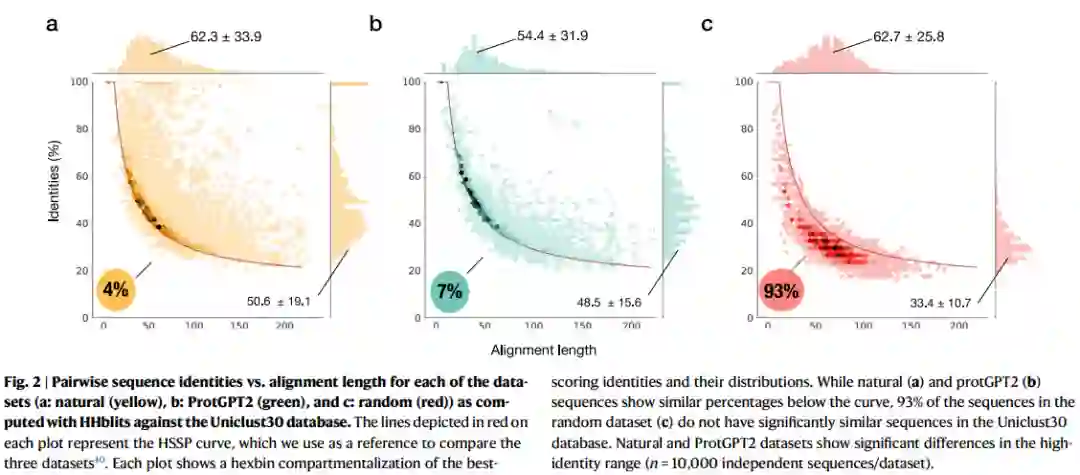
当查看曲线上方和下方的点击分布时,作者观察到HHblits在Uniclust30数据库中发现了许多与自然序列数据集相关的点击(图2a)。具体来说,在10000个数据集序列中,9621个(96.2%)在HSSP曲线以上显示出一致性。类似地,9295个ProtGPT2生成的序列(93%)在Uniclust30数据库中也有对应的序列,它们在HSSP曲线上方对齐(图2b)。相反,93%的随机生成序列低于该阈值(图2c)。尽管natural和ProtGPT2数据集的模式相似,但这两个数据集在点击分布上存在差异。一个标准差范围为31.5–69.7%,自然数据集的平均同一性高于ProtGPT2集,范围为32.9–64.1%(图2a,b)。自然和ProtGPT2序列分布之间的差异无统计学意义(p值90%)。尽管ProtGPT2数据集中的365个序列在Uniclust30中具有高同一性序列,但它们在所有情况下都对应于低于15个氨基酸的比对,而自然数据集显示760个序列超过90%,比对长度在14.8–77.3个氨基酸的一个标准差范围内。这些结果表明,ProtGPT2有效地生成了与自然序列有远亲关系的序列,但不是记忆和重复的结果。
ProtGPT2生成有序结构 设计全新蛋白质序列时最重要的特点就是能否够折叠成稳定的有序结构。作者在AlphaFold预测、Rosetta松弛分数和分子动力学(MD)模拟下,评估了ProtGPT2序列与自然和随机序列的潜在适合度。
AlphaFold在0-100(pLDDT)范围内生成其置信度的每残留估计值。该分数已被证明与顺序相关:低分数(pLDDT>50)往往出现在无序区域,而优秀分数(PLDD>90)出现在有序区域。在这里,作者对每个序列进行了五个结构预测。当采用每个序列的最佳评分结构时,数据集的平均pLDDT为63.2,当对每个序列的所有五个预测进行平均时,为59.6。此外,37%的序列显示pLDDT值超过70。由于pLDDT分数是结构顺序的代理,作者转向自然和随机数据集,看看它们与ProtGPT2序列相比如何。与之前的工作一致,自然数据集中66%的序列预测pLDDT值大于7043,整个数据集的平均值为75.3(补充图2b)。相反,随机数据集中的预测显示pLDDT的平均值为44,pLDDT值超过70的序列中只有7.4%(补充图2c)。
为了进一步验证模型的质量,作者在三个数据集上执行了Rosetta RelaxBB。Rosetta Relax对Rosetta能量函数执行蒙特卡洛优化,从而产生不同的骨架和转子分子构象。较低的罗塞塔能量构象与较松弛的结构相关。最新的罗塞塔能量力场与热容、密度和焓等实验变量密切相关。该评分函数反映了一种静态蛋白质构象的热力学稳定性。在这里,作者对三个数据集的30000个序列进行了Rosetta松弛实验(图3a)。一个广泛的经验法则是,总分(罗塞塔能量单位,REU)应介于−1和−3/残基。作者在天然和ProtGPT2数据集中观察到这种分布,平均值分别为1.90和1.73 REU/残基。正如预期的那样,随机序列的数据集显示平均值为0.13 REU/残基。
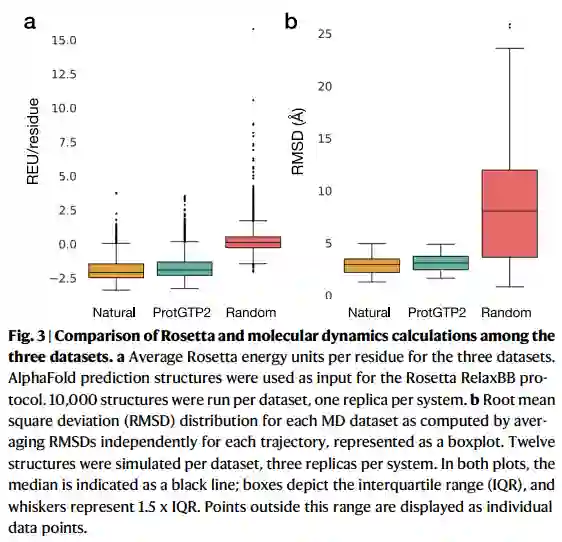
作者进一步测试了ProtGPT2序列是否显示出与自然序列类似的动态特性。蛋白质是动态实体,如果没有其固有的灵活性,它们将无法与其他生物分子相互作用并在细胞中发挥其功能。为了评估ProtGPT2生成的序列是否在与天然蛋白质相同的范围内显示灵活性,作者随机选择每个数据集的12个序列,并运行了100个分子动力学(MD)的三个副本,共108条轨迹,总时间为10.8微秒。为了确保在模拟过程中观察到的动力学不是不同pLDDT值的伪影,因此可能是不同的无序预测,作者确保数据集pLDDT平均值之间的差异在统计上没有差异。自然和ProtGPT2数据集中每条轨迹的均方根偏差平均值分别为2.93和3.12 分别为(图3b)。正如预期的那样,随机序列在轨迹期间显示出显著的偏差,平均值为9.41 Å. 虽然ProtGPT2序列的值高于自然序列,但分布没有显著差异。结果表明,ProtGPT2序列可能与自然界中发现的蛋白质具有类似的动力学特性。
ProtGPT2超越了当前蛋白质空间边界 有几项研究试图将蛋白质序列的维度降低为几个可识别的维度,以便进行分析。大多数表示方法包括(i)蛋白质结构的层次分类,如ECOD和CATH数据库,(ii)笛卡尔表示和相似网络。作者最近在一个网络中表示结构空间,该网络将蛋白质显示为节点,当它们在common中具有同源和结构相似的片段时连接,并在Fuzzle数据库中提供结果。该网络代表来自七个主要SCOP类的25000个域,并表明现代已知的蛋白质空间既有连接的区域,也有“岛状”区域。
进化已经探索了所有可能的蛋白质序列,但并不可信。因此,作者想设计出探索蛋白质空间未探索区域的蛋白质,以及是否可以设计出新的拓扑结构和功能,提出了挑战。作者将ProtGPT2序列整合到作者的蛋白质空间网络表示中。为此,作者为每个SCOPe2.07和ProtGPT2序列生成了HMM配置文件,使用HHsearch以all对all的方式对它们进行比较,并用Protlego表示网络。为了避免具有多个对齐的特定序列最终由网络中的同一节点表示,作者用两个不重叠的对齐复制条目。
该网络包含59612个顶点和427378条边,包括1847个组件或“岛状”簇(图4)。主要成分聚集了一半以上的节点(30690),这一数字明显高于在相同设置下生成的网络中观察到的数量,但不包括ProtGPT2序列,强烈表明ProtGPT2生成的序列桥接了蛋白质空间中的独立岛。作者从拓扑不同的作用域类中选择了跨越网络不同区域的六个示例,以在结构级别展示ProtGPT2序列(图4)。特别是,作者报告了一个全β(751),两个α/β(42661068),一个膜蛋白(4307),一个α + β(486)和全α(785)结构。这些结构说明了ProtGPT2在生成从头结构方面的多功能性。对于每种情况,作者使用FoldSeek58搜索PDB数据库中发现的最相似的蛋白质结构。ProtGPT2生成折叠良好的全β结构(751,4307),尽管最近取得了令人印象深刻的进展,但长期以来一直非常具有挑战性。ProtGPT2还生成膜蛋白(4307),由于在指定膜内结构方面的挑战和繁重的实验表征,这对蛋白质设计提出了一个困难的目标。除了生成自然折叠代表外,ProtGPT2还产生了以前未收录的拓扑结构。例如,作者报告了蛋白质4266,其拓扑结构与目前PDB收录中的任何结构都不匹配,DALI Z分数低为5.4,RMSD为3.0 Å到PDB 5B48超过67个残基(9%)。
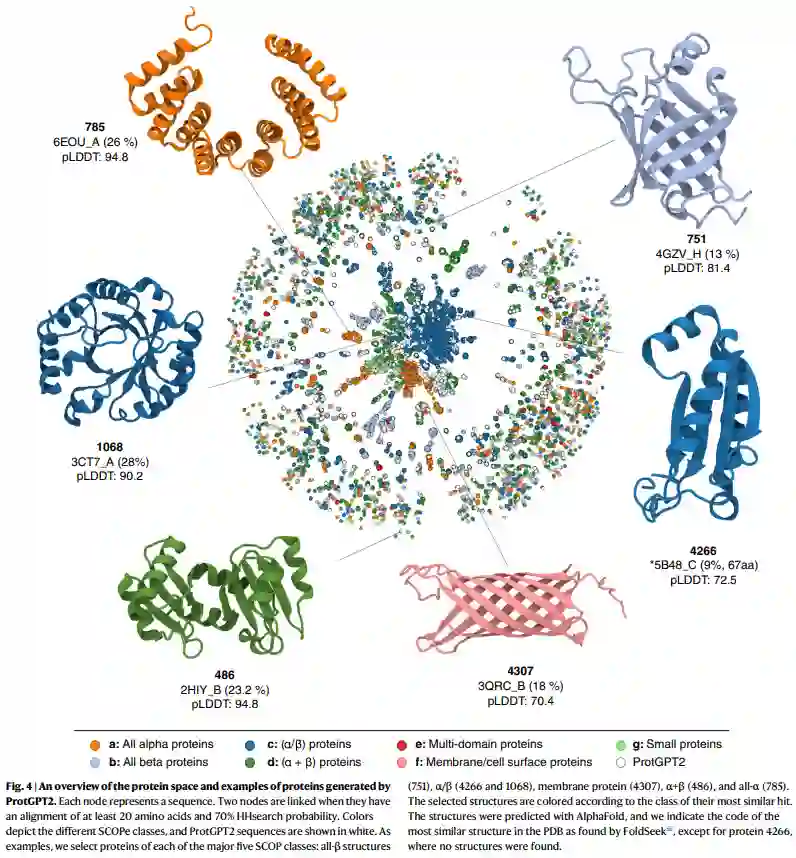
然而,ProtGPT2序列最显著的特性可能是其与所有先前设计的从头结构的显著偏差,这些结构通常具有环路和最小结构元素的理想拓扑。从头蛋白质设计的优点是不携带任何进化历史,但在实践中,缺乏实例和更长的环阻碍了与其他分子相互作用和功能实现所需的裂缝、表面和空腔的设计。ProtGPT2序列类似于天然蛋白质的复杂性,具有多方面的表面,能够分配相互作用的分子和底物,从而为功能化铺平了道路。在图4中,作者展示了结构486和1060,这是此类复杂结构的两个示例。特别是,1068显示了TIM-barrel褶皱,该拓扑迄今为止在从头蛋白质设计中取得了成功,但其理想化结构已被证明具有挑战性,无法通过额外的secondary elements和更长的环进行扩展。
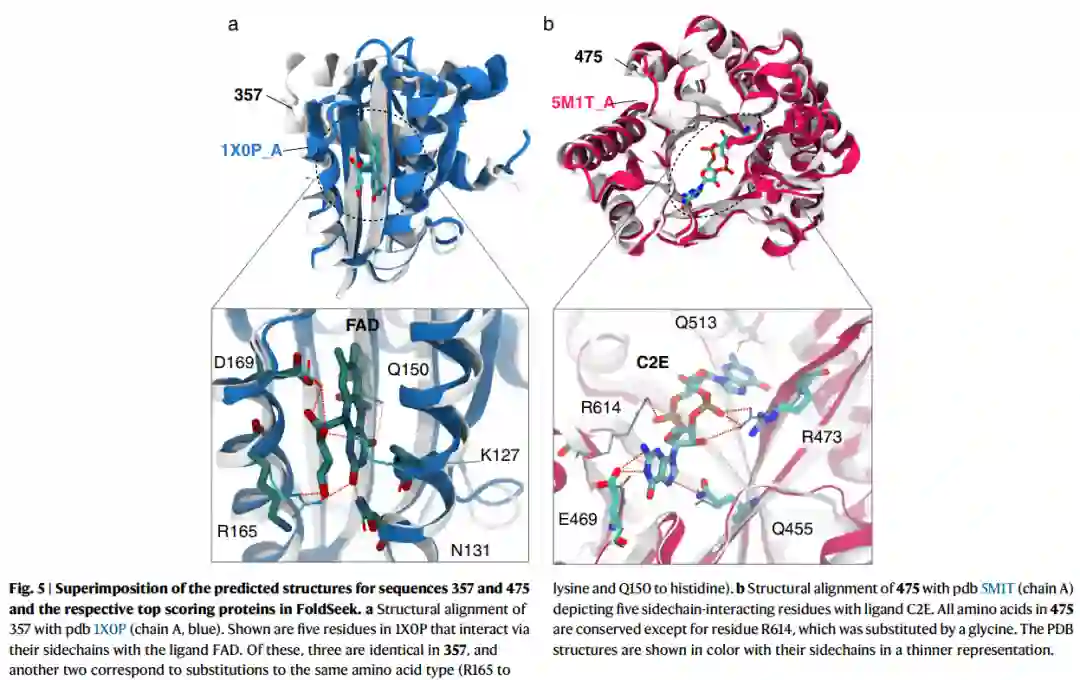
保留的功能热点 对FoldSeek发现的最佳点击结构叠加进行检查,发现了几个配体相互作用残基的侧链保守的例子。图5中显示了两个示例。最类似于序列357(图5a)的自然结构对应于PDB代码1X0P(链A),一个结合FAD的蓝光传感器域。当重叠结构时,作者观察到357保留了侧链结合热点,三个残基相同(D169、Q150和N131),两个不同但能够形成相同相互作用的残基,即R165位的赖氨酸和K127位的组氨酸。序列475(图5b)最类似于PDB代码5M1T(链A),折叠成TIM-barrel并与细菌第二信使环二-3′,5′-鸟苷单磷酸(PDB三字母代码C2E)结合的磷酸二酯酶。在五个侧链相互作用的残基中,ProtGPT2序列保留了三个残基(Q455、R473和E469),并包括一个取代另一个能够氢键的残基(用于Q513的天冬氨酸)。值得注意的是,ProtGPT2以zero-shot生成了这些序列,即在这两个特定折叠中没有进一步微调。这些结果对蛋白质工程产生了重大影响,因为尽管同源性较低(357和45分别为31.1%和29.2%),但ProtGPT2似乎保留了生成序列中的结合位置,并可用于增加特定折叠和家族的序列。
4 总结 过去的2年里,利用人工智能方法的从头蛋白质设计取得了惊人的成功。作者构建了一个自回归语言模型ProtGPT2,ProtGPT2有效地学习了蛋白质语言。ProtGPT2可以生成与自然序列有远近关系的序列,其结构类似于已知的结构空间,具有非理想化的复杂结构。由于ProtGPT2已在整个序列空间上训练,因此该模型产生的序列可以对任何区域进行采样,包括深色蛋白质组和传统上认为在蛋白质设计领域非常具有挑战性的区域,例如全β结构和膜蛋白。ProtGPT2蛋白质与远亲天然蛋白质结构的视觉叠加显示,PROTGPT1还捕获了功能决定簇,保留了配体结合的相互作用。由于人工蛋白质的设计可以解决许多生物医学和环境问题,作者在蛋白质语言模型中看到了巨大的潜力。ProtGPT2设计在几秒钟内即可适应球状蛋白质,无需进行进一步训练。通过根据用户序列微调模型,ProtGPT2可以适应特定的家族、功能或折叠。在这种情况下,ProtGPT2将能够筛选与天然蛋白质相似的蛋白质,以改善、微调或改变天然蛋白质的特定生化功能。大规模筛选ProtGPT2设计的蛋白质库可能会识别数据库中未捕获的蛋白质折叠以及在自然界中没有相关对应功能的蛋白质。ProtGPT2是面向高效蛋白质设计和生成迈出的一大步,为探索设计蛋白质结构和功能的参数及其后续实际应用的实验研究奠定了基础。 参考资料 Ferruz, N., Schmidt, S. & Höcker, B. ProtGPT2 is a deep unsupervised language model for protein design. Nat Commun 13, 4348 (2022). https://doi.org/10.1038/s41467-022-32007-7
项目主页 https://huggingface.co/nferruz/ProtGPT2
代码 https://huggingface.co/docs/transformers/main_classes/trainer
