综述:使用语言模型进行可控的蛋白质设计
编译 | 白菜叶

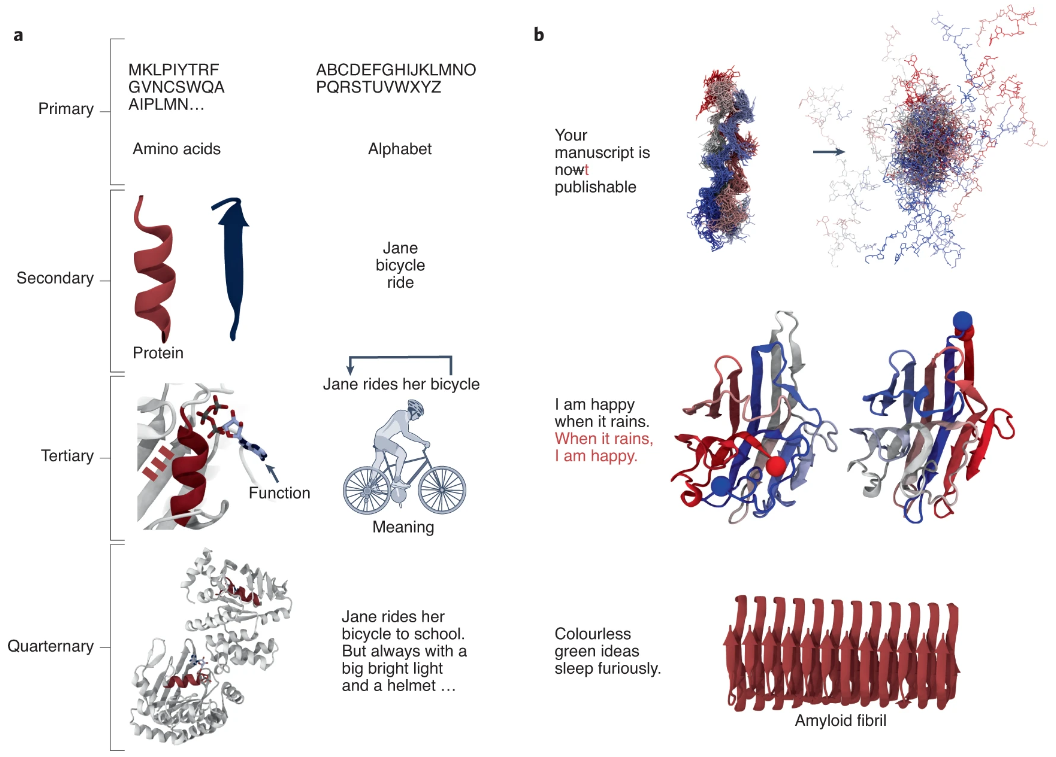
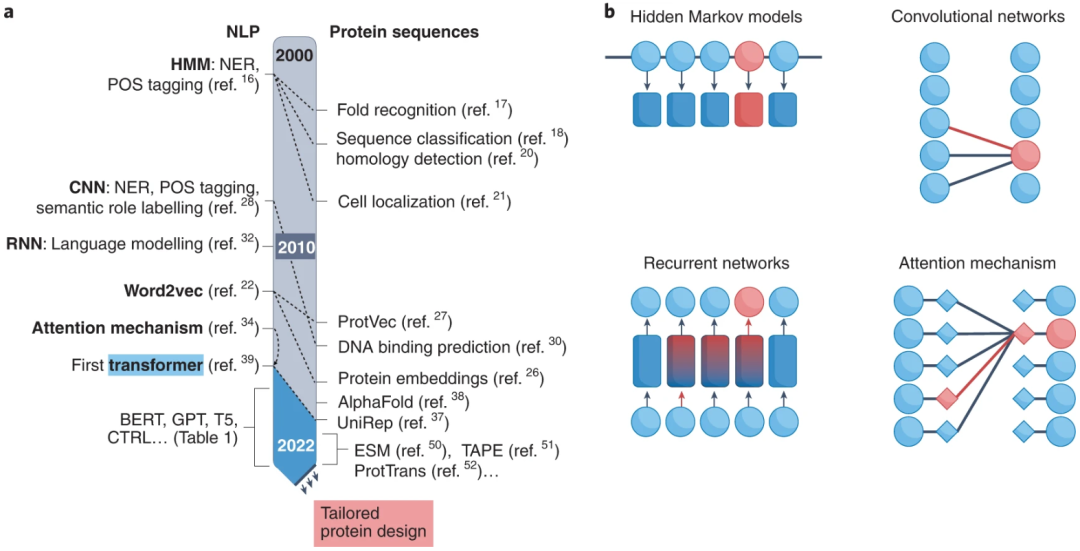
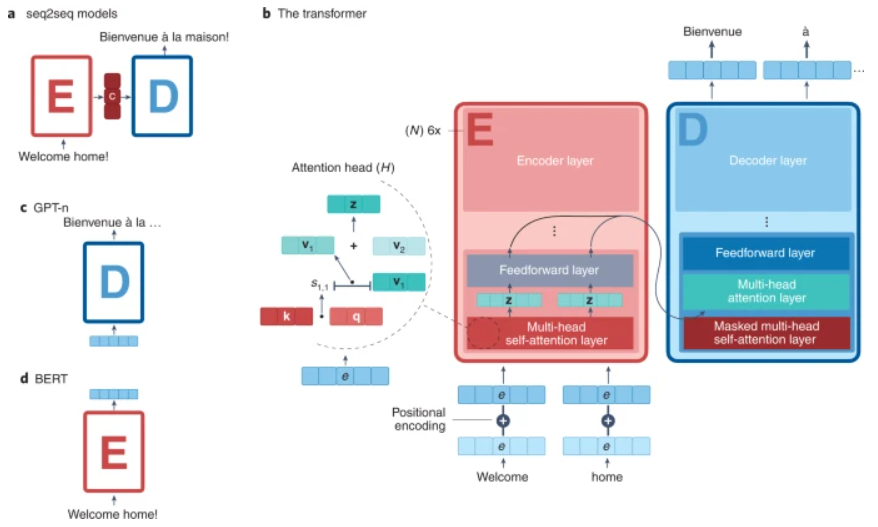
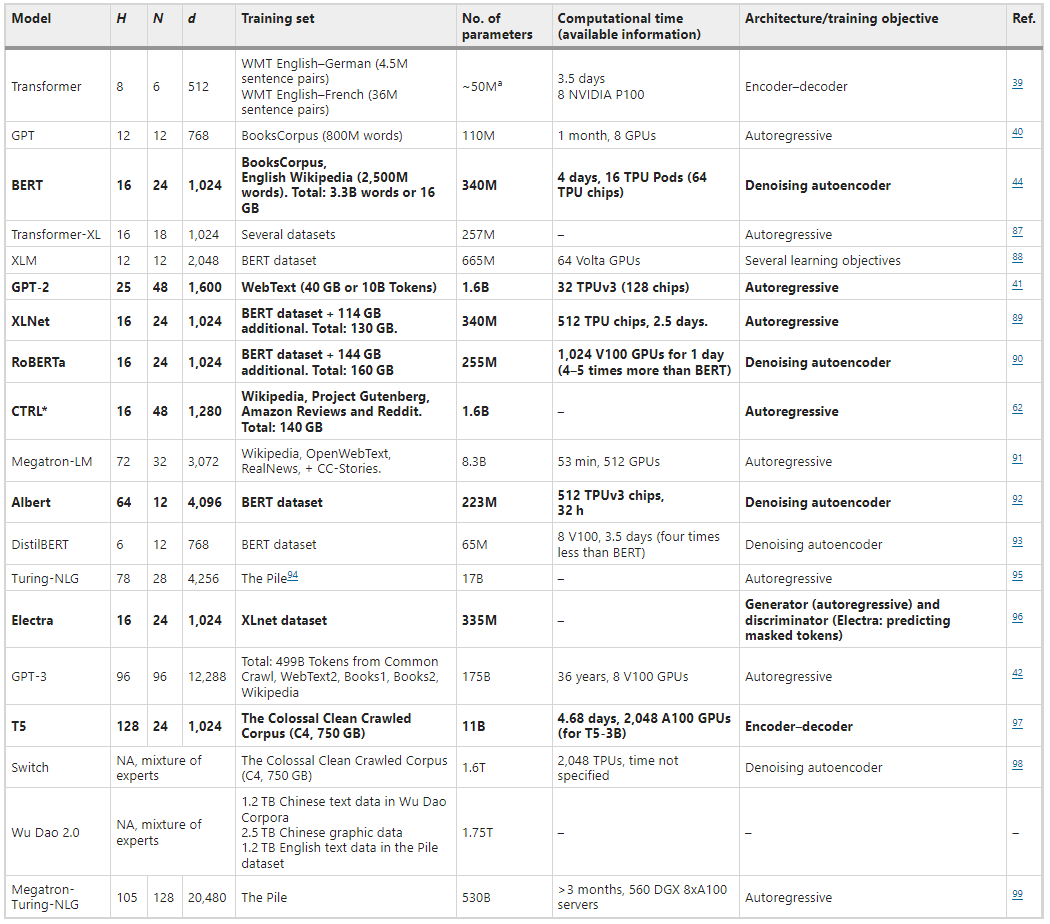
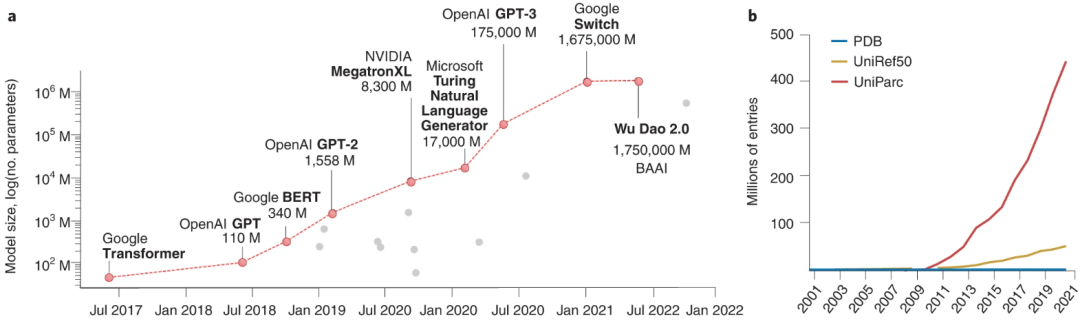
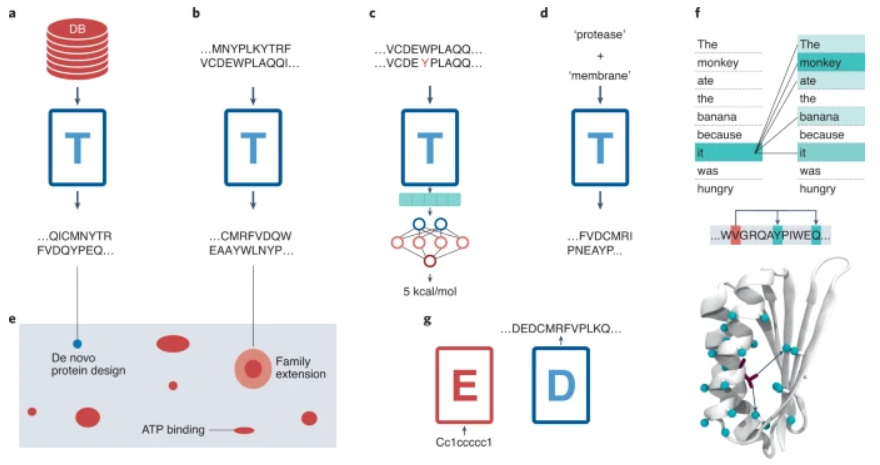
图示:蛋白质和语言之间的相似性。(来源:论文)
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。
登录查看更多
相关内容
专知会员服务
67+阅读 · 2022年4月15日
Arxiv
0+阅读 · 2022年9月15日
UniViLM: A Unified Video and Language Pre-Training Model for Multimodal Understanding and Generation
Arxiv
19+阅读 · 2020年2月15日
相关VIP内容
专知会员服务
67+阅读 · 2022年4月15日
相关资讯
相关论文
Arxiv
0+阅读 · 2022年9月15日
UniViLM: A Unified Video and Language Pre-Training Model for Multimodal Understanding and Generation
Arxiv
19+阅读 · 2020年2月15日