编译|程昭龙
审稿|林荣鑫,王静本文介绍由不列颠哥伦比亚大学的Yongjin P. Park通讯预印在bioRxiv的研究成果:在多细胞生物中,细胞特性和功能是通过与周围其他细胞的相互作用来启动和完善的。在此,**作者提出了一种名为SPURCE的可扩展机器学习方法,旨在系统地确定嵌入单细胞RNA序列数据中常见细胞间的通信模式。**作者将该方法应用于研究肿瘤微环境,并整合了多个乳腺癌数据集,发现了七个经常观察到的相互作用特征和潜在的基因-基因相互作用网络。实验结果表明,通过不同的相互作用模式而不是已知标记基因的静态表达,可以更好地理解肿瘤异质性的一部分,尤其是同一亚型内的肿瘤异质性。
1 简介 单细胞RNA测序(scRNA-seq)技术的发展已成为基因组学的新前沿。在单细胞分辨率下进行多模态组学定量分析,有助于深入了解癌症生物学的不同方面。癌症研究的基本问题之一是癌细胞如何在肿瘤微环境(TME)等受限的异质环境中相互作用,研究表明,TME中细胞群之间的细胞间通信(CCC)在肿瘤生长和转移过程中至关重要。了解肿瘤及其相互作用的合作细胞之间的复杂通信可以帮助确定潜在的癌症治疗途径。
要理解TME中细胞间相互作用的一个重大技术挑战就是需设计一种系统化的方法,从每个相互作用的细胞对中分离和捕获相互作用信号。研究CCC的传统方法包括低维空间中的聚类特征和推断已知细胞类型簇之间的相互作用。虽然这些方法揭示了许多控制细胞分化和发病的信号机制,但它们假设每个簇(使用有限数量的标记基因注释)代表一种细胞类型,集群中的所有细胞都以相同的方式相互作用。因此,这些方法不能解释簇内细胞的异质性。同一细胞类型内的细胞可能存在多种亚型/状态,并根据相互作用的合作细胞的类型和状态表现出不同的相互作用模式,这对于理解癌症进展至关重要。此外,不同环境(例如疾病状态)下的细胞之间的相互作用是单独进行研究的,这会丢失特定环境的可变性信息,并且重复性强,计算成本高。
最近的研究已经解决了这些挑战,并开发了捕捉同一簇内细胞相互作用多样性的方法,例如Tensor-cell2cell、scTensor等。然而,这些方法依赖于细胞类型和聚集细胞的先验知识,根据配体-受体(LR)基因的平均表达来计算通信分数。
因此,作者提出了一种新的计算方法SPRUCE,通过复合嵌入解开单细胞成对关系,以可扩展的方式分析数千万个细胞对。采用已知的配体和受体蛋白质-蛋白质相互作用,作者研究了细胞对定位在潜在主题空间附近的原因和方式,并强调了在肿瘤微环境数据中反复观察到的常见模式。SPRUCE基于嵌入式主题模型(ETM),ETM是一种基于变分自动编码器架构的生成性深度学习方法,并使用可解释的主题特定基因表达式字典矩阵表示低维主题空间中的单细胞矢量数据。它已经在自然语言处理中成功实现,可以提取代表大规模文档的有意义主题,最近一项名为scETM的研究表明,基于ETM的技术可以有效地从稀疏和异质的单细胞数据中捕获重要的生物信号。SPRUCE的关键贡献是通过表征LR基因驱动的细胞-细胞相互作用模式,在多个数据集中无偏地识别可解释的细胞亚型/状态。现有的基于图的单细胞分析方法通常将细胞间交互模块定义为图中紧密相连的组件(邻接矩阵)。作者提出的SPRUCE模型将细胞间相互作用模式视为边缘流,或一个巨大的关联矩阵。
2 结果 乳腺癌研究中SPRUCE模型训练概述
作者结合现有的乳腺癌数据集和癌症特异性免疫细胞数据,为一项无偏乳腺癌研究构建了一个全面的单细胞目录,产生了一个由20288个基因和155913个细胞组成的数据矩阵。作者首先将来自多个数据集的细胞映射到一个共同的潜在主题空间(K=50),并通过基于变分自动编码器的主题建模来对它们进行协调,而不是提出额外的假设。然后,基于细胞间潜在主题空间的余弦相似度,构建细胞间交互网络并进行分层抽样,使主题-主题关系在SPRUCE训练的后续步骤中以相似的方式表示。对于每一个25M+的细胞对,作者提取了已知的648种配体蛋白和672种受体蛋白的基因表达,并将其用作SPRUCE模型训练的特征载体。
多项式概率主题模型确定了11种已知细胞类型的50个细胞主题
作者使用贝叶斯深度学习方法来估计包含50个潜在维度的155913个细胞的嵌入主题模型。研究发现每个细胞主题对应一组平均数量为3118个细胞 (图1A)。在50个主题中,包含100个以上细胞的主题有32个(图1C)。将最多数量的细胞(占数据集的24%)分配给主题37,其中96%的细胞是之前确定的免疫细胞(T细胞和B细胞)。数据集中98%的癌细胞被分配到13个细胞主题,这13个主题中9个主题的癌细胞比例大于95%。通过使用UMAP对标注了细胞类型的潜在细胞主题进行可视化,显示每个主题的不同簇,其中大多数细胞是数据集中的主要细胞类型之一(图1B)。同时,作者进一步证实了与之前使用细胞类型谱系典型标记进行分析的一致性(图1D)。估计的细胞主题比例表明常驻细胞类型具有相似的主题比例。然而,癌细胞具有不同的混合主题比例,这表明该模型识别了癌细胞的许多不同主题(图1E)。作者还尝试了10个、25个和50个不同数量的细胞主题,并决定使用具有50个细胞主题的主题模型,因为该模型对主要细胞类型,尤其是癌细胞,显示出了分离良好的不同簇。
图1 概率主题模型为细胞类型确定细胞主题
在2500万个细胞对中发现7个稳定的TME特异性相互作用特征
作者用来自155913个细胞的LR基因表达数据来构建24790167个细胞对,并估计具有25个潜在维度的嵌入交互主题模型,同时,通过SPRUCE模型可确定常见的交互模式(图2A,B)。在代表2500万个细胞对的25个交互主题中,7个主题占总细胞对交互的55%,每个主题包含3%+的细胞对(图2C)。其他18个交互主题,每个主题占总细胞对交互的2%,嵌入了基线交互信号。
该模型估计了每个交互主题中的LR基因负载,这些交互主题描述了每个基因的相对贡献,实验中可以对这些负荷进行排序,从而在每个交互主题中识别生物学上可解释的主题特异性顶部基因(图2D)。在主要相互作用主题中,排名靠前的LR基因富集了不同细胞类型特异性的功能相互作用。为了确认每个交互主题都捕获了细胞类型特异性CCC,作者仔细研究了数据集中所有细胞在每个交互主题中目标细胞的细胞类型分布。结果发现,在每个相互作用主题中,富集的顶部LR基因的功能作用与该主题中靶细胞的主要细胞类型匹配(图2E)。此外,交互主题在各细胞类型中的分布进一步证实了交互主题的细胞类型特异性富集。与髓样细胞、T细胞、内皮细胞、CAF和PVL细胞类型相比,癌细胞、上皮细胞、血浆细胞和B细胞显示出异质性的相互作用模式(图2F)。对于癌细胞而言,大多数相互作用属于癌症生长和癌症转移的主题,其中许多排名靠前的基因是癌基因。

图2 SPRUCE模型概述及确定的交互模式
交互主题提供了一种理解乳腺癌异质性的方法
接下来,作者基于由细胞主题模型捕获的无偏转录组特征来研究乳腺癌细胞的异质性,同时将细胞主题与SPRUCE分析表征的相互作用主题相关联(图3A)。25835个癌细胞的相互作用模式表现出了所有的相互作用模式(图3B)。细胞主题模型确定了癌细胞的13个细胞主题,显示了与其靶细胞相互作用的独特模式(图3C)。正如预期的那样,癌症生长主题在 13 个细胞主题的 7 个中占主导地位。
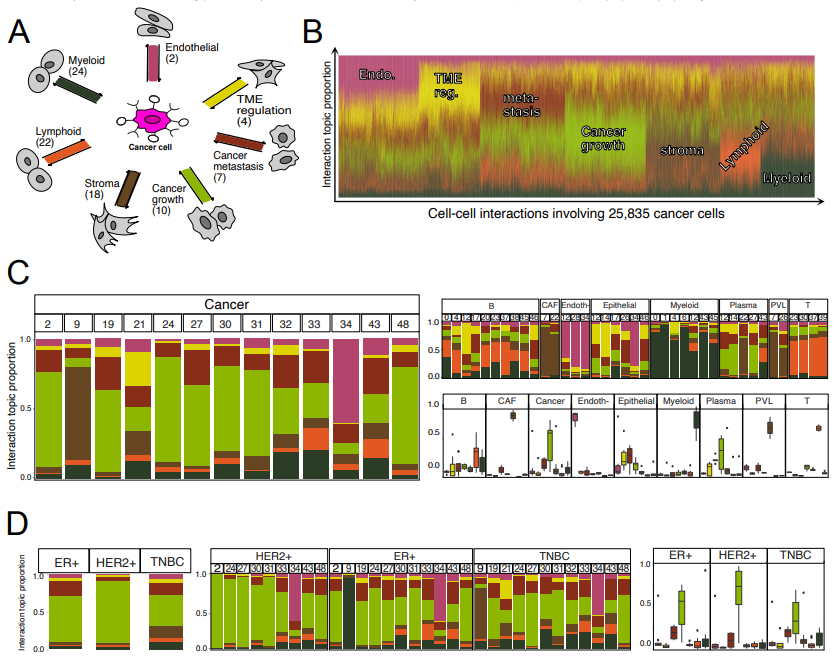
图3 细胞主题特异性相互作用模式揭示癌细胞的异质性
乳腺癌细胞根据疾病的基因组学和病理学分为不同的亚型,不同的亚型通常会导致明显不同的临床结果。所有三种不同的癌细胞亚型表现出了不同的相互作用模式,其中 TNBC(三阴性乳腺癌)细胞的异质性与 HER2+ 和 ER+ 亚型相比更高(图 3D)。此外,SPRUCE在这些癌症亚型中确定了显示特定主题相互作用模式的特定细胞群(细胞主题)。
交互主题揭示潜在的癌症亚型特异性基因相互作用网络
SPRUCE还揭示了模型参数矩阵中特定主题的相互作用模式(图4),通过该模式可以估计基因-基因相关网络(配体与受体)。同时,与癌细胞相邻的相互作用网络通常包括已知的癌症相关基因和其他参与细胞发育过程的基因。通过分析将它们放在相同的网络模块中,这些相互作用对于癌细胞控制正常细胞过程的潜在作用。此外,在淋巴主题中,T细胞受体复合物和TCR信号通路基因在癌症和周围免疫细胞中均高表达。其他趋化因子受体基因,如CXCR3和CXCR4,也被发现在这一相互作用主题中高度共激活,这证实了T细胞和癌细胞之间的交互作用在促进癌症生长、免疫逃避和转移中的关键作用。
图4 交互主题构建基因网络
3 总结 在这项研究中,作者提出了一种新颖的机器学习框架,该框架系统地剖析了数千万个细胞对,并揭示了关于细胞表面配体-受体蛋白相互作用的细胞相互交流的共同模式。特别是,作者的分析侧重于通过重新分析最先进的单细胞基因组学数据集来寻找乳腺癌进展和转移中常用的沟通渠道。SPRUCE建立在概率主题模型和变分自动编码器模型的基础上,具体地证明了癌症异质性的一部分可以在癌症细胞的不同和特定环境的相互作用中理解。实验还发现了许多配体-受体相互作用可以以一种亚型特异性的方式发生,尽管在传统的单细胞分析中,细胞主要聚集为癌细胞。在作者的研究结果中,细胞类型和状态得到了更好的理解,并且在考虑细胞间通信模式的同时,可以细化细胞类型的定义。
SPRUCE概括了现有的生物信息学方法,并且不依赖于规定的细胞类型注释/聚类结果,这可能会在下游分析中引入不必要的偏差。此外,如果簇内的细胞不像预期的那样均匀,则基于簇的细胞-细胞交换方法很容易导致混淆相关统计,明显违反必要的假设,如独立和识别分布的表达值。对于多组学数据集成任务,可以通过连接多个数据模式来研究多模态表达的共现,例如DNA可及性、组蛋白修饰和代谢组学。
SPRUCE方法依赖于几个专门的建模假设。其中之一是假设已知的配体-受体蛋白质-蛋白质相互作用网络充当主题特异性相互作用网络的超集/主干。考虑到大多数蛋白质-蛋白质相互作用是通过易出错的高通量方法在体外实验中发现的,因此在相互作用分析方法的精确度和特异性方面仍有改进的空间。在这里,作者只关注了直接的表面蛋白相互作用。然而,确定来自表面蛋白单链通路的下游基因的因果效应可以进一步丰富对疾病病因的理解。
参考资料 Subedi S, Park Y. SPRUCE: Single-cell Pairwise Relationships Untangled by Composite Embedding model[J]. bioRxiv, 2022.