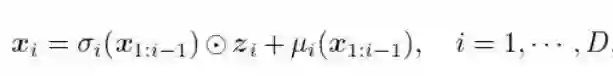今天给大家介绍一篇来自于icml2022关于在特定结合位点生成目标3D分子的文章,本文的标题是《Generating 3D Molecules for Target Protein Binding》。

1.背景
首先本文提出了在当现存的生成与蛋白质结合的分子存在的问题,一共有三点。第一,现存的很多方法忽视了复杂的条件信息包含3d空间信息以及原子之间的化学信息;第二,本文认为应该能够在连续的3d空间放置生成的分子,而此前的方法满足不了这一点,即只能在离散化的空间放置生成的分子;第三,现在的方法,无法保证在蛋白质的环境发生变化的情况下(发生了相关的平移以及旋转),生成对应的分子也随之发生相应的改变(进行相关的平移和旋转)。如果环境发生改变(发生了相关的平移以及旋转)生成对应的分子也随之改变(进行相关的平移和旋转),这种特性称为等变性。第三点换句话就是无法保证分子的等变性。 本文提出的这个框架(GraphBP)可以有效的解决上述的三个问题,GraphBP通过将特定类型和位置的原子一个一个地放置到给定的结合位点来生成与给定蛋白质结合的 3D 分子。在每一步,首先使用 3D 图神经网络从中间上下文信息中获取几何和化学信息表示。上下文包括给定的结合位点和在前面步骤中放置的所有原子。其次,为了保持理想的等变性质,该框架根据设计的辅助分类器选择一个局部参考原子,然后构建一个局部球坐标系。最后,为了放置一个新原子,首先生成原子类型和相对位置 。文章通过流动模型构建局部坐标系。同时文章中对于原子的种类以及相对位置提出了一种按顺序生成变量的方法(通过原子种类a生成一部分位置信息d再通过原子种类a和一部分位置信息d得到该原子其他的位置信息θ,最后通过a、d、θ得到该原子最后剩余的位置信息φ),并通过实验验证了这种依赖关系对于生成3D分子的有效性以及可靠性。实验表明,GraphBP 可有效生成具有与目标蛋白质结合位点结合能力的 3D 分子。
2.方法
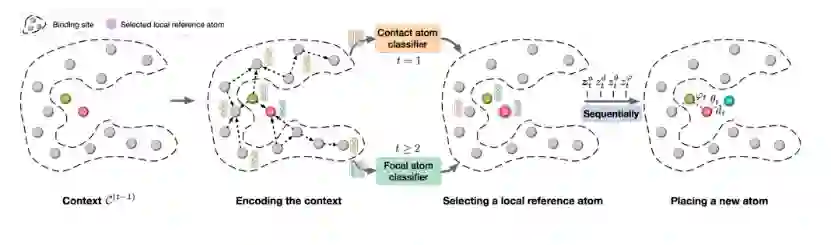
本文中生成分子的过程如图1所示:
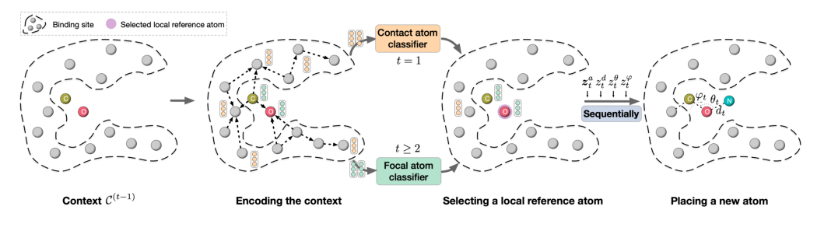
图1.GraphBP生成分子示意图
第一步,编码上下文信息。本文首先将各个原子之间进行连线构成一张Graph。然后将这种图作为输入放入3DGNN的网络中,为每个分子生成对应的信息。在3DGNN中存在一个重要的输入变量即每个原子与其临近原子的距离d,而这个距离不会因为位置的平移和旋转发生改变,所以每个原子的通过3DGNN获得的编码信息不会发生改变。 第二步,通过原子分类器选择局部参考原子,构造局部球面坐标系(scs)。首先第一步使用contact atom classifier(接触原子分类器)对结合位点的原子进行选择(第一步只存在结合位点的原子,没有配体原子)。contact atom classifier(接触原子分类器)将结合位点的所有原子的编码作为输入,进行选择,选择出最佳的局部参考原子(结合位点中离配体最近的原子)。之后的步骤通过focal atom classifier(焦点原子分类器),从配体中的所有原子进行选择,选择出焦点原子作为局部参考原子。我们需要空间的三个点去定义scs,假设选择的局部参考原子点是上下文c(t-1)中f原子,需要我们在c(t-1)选择出离f最近的一个原子c以及第二近的原子e。存在了f,c,e原子,就可以构建scs,同时可以生成该局部球面坐标系的三元组(dt,θt,φt)。dt是生成原子t和f原子的距离,θt是生成原子t与f原子之间的线段和c原子和e原子之间线段之间的夹角,φt是生成原子t ,f原子 ,c原子的平面和e原子,f原子, c原子的平面 第三步,放置新的原子,通过一个Autoregressive flow models(自回归流模型)得到生成原子的类型和三元组。首先,flow(流)模型表示经过一个参数化的可逆变换函数fθ:Z ∈ RD → X∈ RD,将服从某个先验分布的隐变量Z变换到另一个变量X。Autoregressive flow models(自回归流模型)是一种特定的flow(流)模型,其中变换函数被表述为Autoregressive models(自回归模型)即X的每一维Xi的生成都以该维前面的每一维X1:i-1作为条件,如下;
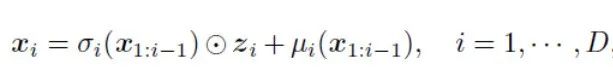
t代表第t步产生的原子,本文按照at→dt→θt→φt的顺序来添加原子即存在如下的依赖关系C(t−1) → at, (C(t−1), at) →dt, (C(t−1), at , dt) →θt, (C(t−1), at, dt ,θt) → φt,同时在每一步的生成过程添加 zta,ztd,ztθ,ztφ flow (流)模型的隐变量。在生成过程中,从已知的先验高斯分布中采样对应的隐变量z,然后将相对应的隐变量z和依赖关系通过g函数(自回归函数)映射到at与dt, θt, φt 即ga(C(t−1),zta) → at,gd(C(t−1),ztd ,at ) → dt,gθ(C(t−1),ztθ,at, dt) → θt ,gφ(C(t−1),ztφ,at, dt ,θt) → φt ,在训练过程中由于at是离散值,不满足流模型的条件,需要通过添加高斯噪音将其连续化。,同时将获得的进行argmax的操作获得one-hot码at 。g的表达如图2,每次将上下文信息和依赖关系作为输入,依次得到原子种类和三元组(dt,θt,φt)当得到最后的三元组(dt,θt,φt),就可以通过之前建立的局部球面坐标系(scs)放置新的原子。 总的来说,文章通过参考原子,建立局部球面坐标系,生成坐标三元组,一方面保障了生成原子的等变性(平移不变和旋转不变)同时所有步骤中的生成原子构成生成分子,进而保障了生成分子的等变的特性。
3.训练
在文章中涉及到三个损失函数:1.Atom placement loss Lap2.Contact atom classifier loss Lcc3.Contact atom classifier loss Lcc1采用流模型计算出训练数据的对数似然值,然后取其相反数;2和3损失函数使用的是传统的交叉熵损失,对于2来说结合位点离配体最近的点作为正样本,最远的作为负样本。对于3来说所有原子没有可以连接的位置作为负样本,存在连接的位置作为正样本。同时采用了CrossDocked2020 dataset。** ******
4.结果对比
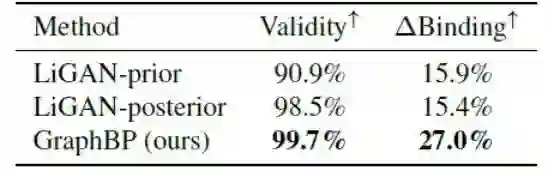
利用两个指标衡量模型的生成性能即(i)Validity(有效性)是指在所有生成分子中化学有效分子的百分比。如果一个分子可以被 RDkit 消毒那么它就是有效的。(ii) ΔBinding(结合性) 衡量生成具有比其相应参考分子更高结合性的分子的百分比。生成模型与LiGAN-prior和LiGAN-posterior进行模型对比,本文中的模型效果非常显著如表1。

表1.不同模型对于药物设计的生成性能,↑表示更好的性能
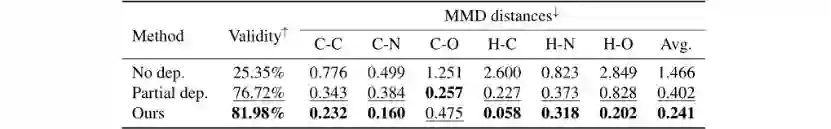
验证之前所提出的按顺序去生成原子类型at和三元组(dt,θt,φt)即at→dt →θt→φt 文章中与完全依赖相对应引入了两种方式进行对比: 1.无依赖 C(t−1) → at, C(t−1) → dt,C(t−1) → θt, C(t−1) → φt 2.部分依赖 C(t−1) → at, (C(t−1), at) → dt, (C(t−1), at) → θt, (C(t−1), at) → φt 比较两个指标,一个是 Validity(有效性)另外一个是生成的3D分子和训练的3D分子之间的键长分布的最大平均差异距离(MMD distances)如表2。
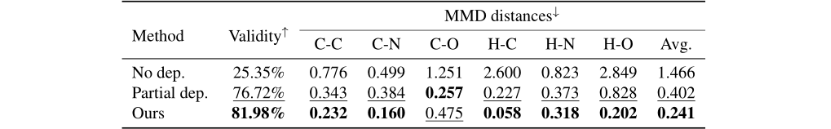
表2.GraphBP和和消融模型在随机分子几何生成任务上的比较,↑(↓)表示数值越高(越低)性能越好
5.最终结论
在本篇论文中,提出了一种机器学习方法(GraphBP)来生成用于靶蛋白结合的3D分子。GraphBP能够捕捉蛋白质-配体复合物的三维几何结构和之间的相互作用,在不离散化三维空间的情况下放置原子,并在生成过程中保持等变特性,GraphBP被证明效果是非常优异的。