ICLR 2022|迭代精调的GNN用于抗体-序列结构协同设计
作者提出了一个生成模型来自动设计具有增强的结合特异性或中和能力的抗体CDR,以前生成方法将蛋白质设计描述为一项结构制约的序列生成任务,前提是预先给定所需的3D结构。相比之下,作者将CDR的序列和3D结构设计为图。该模型以自回归的方式分解一个序列,同时迭代地精调其预测的全局结构。推断出结构反过来指导后续的残基的选择。
为了提高效率,我们以粗粒度的方式对CDR内部和外部的残基之间进行条件独立的建模。作者的模型在测试集上实现了更高的对数似然,在新的抗体上预测、CDR结构预测、抗体-抗原复合物的现有抗体设计、在设计能够中和SARS-CoV-2病毒的抗体方面均优于之前的基线模型。

作者youtube视频
CDR生成中有三个关键的建模问题:第一问题:如何建模序列与其底层3D结构之间的关系。生成没有相应结构的序列可能会导致次优性能,而从预定义的3D结构生成的序列不适用于抗体,因为所需结构在先验中很少已知。因此,开发协同设计序列和结构的模型至关重要。
第二问题是,在给定序列剩余部分(context)的情况下,如何对CDR的条件分布进行建模。基于注意力的方法只在序列水平上模拟条件独立,但CDR与其上下残基之间的结构交互对生成至关重要。
最后一个问题与模型优化各种属性的能力有关。传统的基于物理的方法侧重于结合能最小化,但在实践中,我们的目标可能比结合能更复杂。
作者将序列结构对表示为一个图,并将协同设计任务表示为一个图生成问题。图表示允许我们在序列和结构级别上对CDR与其上下context之间条件独立的进行建模。
之前的自回归模型无法修改生成的结构,因为它们是在techer forcing的情况下训练的。因此在之前的步骤中犯的错误可能会导致后续生成步骤中的一连串错误。为了解决这些问题,作者提出了一种新的结构,将氨基酸节点的生成与三维结构的预测交织在一起。结构生成是基于对全局图的迭代精调,而不是在techer forcing下对局部图进行顺序展开。
由于上下文序列很长,进一步通过将节点分组到块中来引入粗化图表示。在更粗糙的层次上应用图卷积来有效地将上下文信息传播到CDR残基。在已知结构的抗体进行预训练后,我们使用预定义的属性预测器对其进行微调,以生成具有特定属性的抗体。
2 Related Work
抗体/蛋白设计
目前计算抗体设计的方法大致分为两类:一类是基于能量函数优化,使用蒙特卡洛模拟迭代修改序列及其结构,直到达到局部能量最小值。类似的方法也用于蛋白质设计。尽管如此,这些基于物理的方法在计算上是昂贵的,我们预测的目标可能比低结合能要更复杂得多。
第二类是基于生成模型。对于抗体,他们大多是基于序列的。对于蛋白质,进一步发展了以骨架结构或蛋白质折叠为条件的模型。我们的模型还试图结合3D结构信息来生成抗体。由于新病原体通常不知道最佳的CDR结构,因此我们协同设计特定特性的序列和结构。
图生成模型
图生成模型,与图生成的自回归模型有关。特别是Gebauer等人[5]开发了用于分子图和构象协同设计的G-SchNet。与他们按顺序生成边,并且在新节点到达时无法修改先前生成的子图。虽然Graphite[2]也使用迭代细化来预测图的邻接矩阵,但它假设所有节点标签都给定,并且只预测边。相比之下,作者将自回归模型与迭代细化相结合,以生成包含节点和边标签的完整图。
作者的迭代优化模型还与分子构象预测的分数匹配方法和点云的基于扩散的方法有关,这些算法还可以迭代优化预测的3D结构,但只适用于完整的分子或点云。相比之下,我们的方法学学习预测不完整图的三维结构,并将三维结构细化与图生成交叉。
3 Antibody Sequence and Structure Co-Design
概述
抗体的基本单位是残基,其值可以是20个氨基酸中的一个:
每个残基都是一个“word/token“;每个原子都有一个3D坐标。
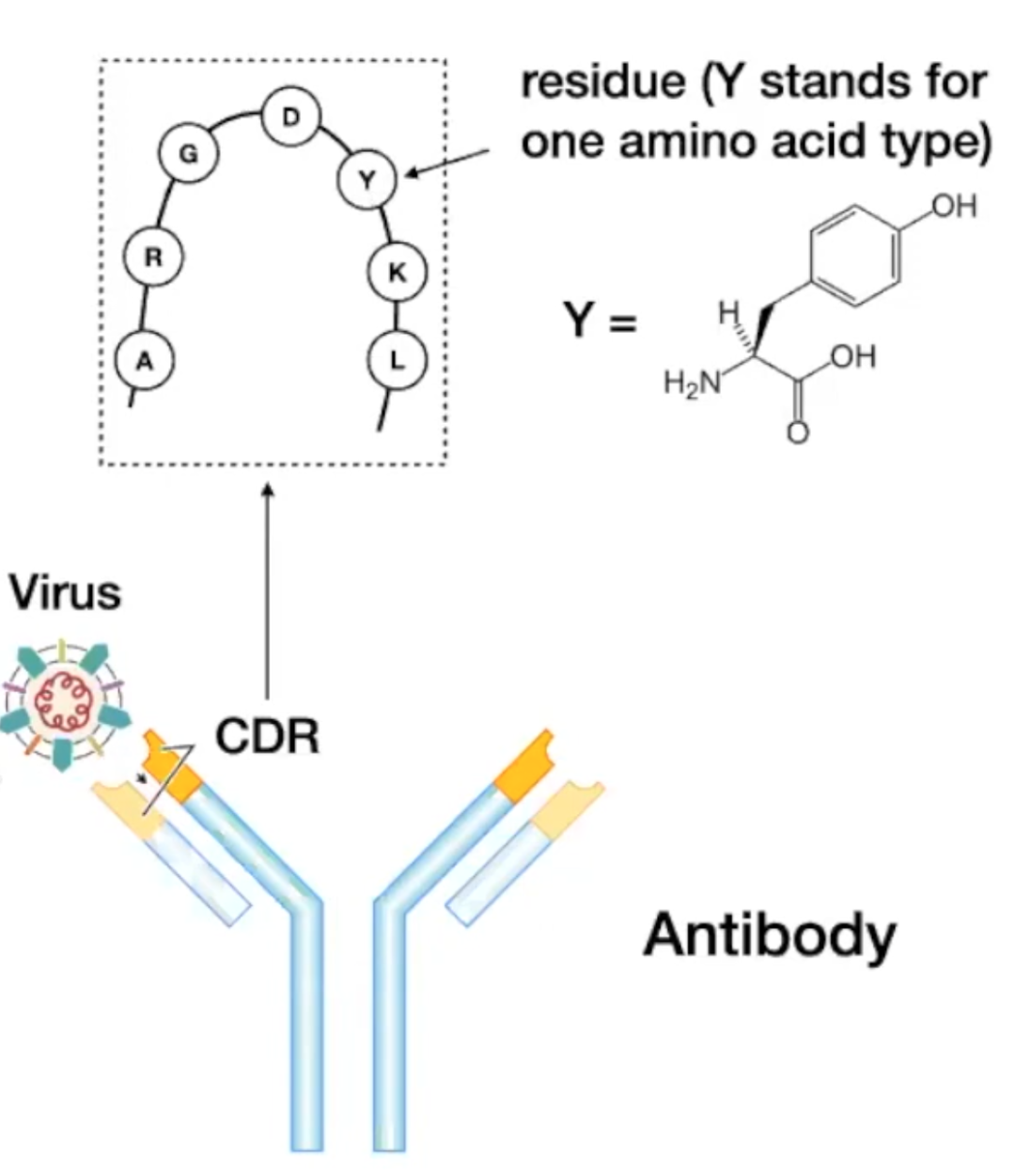
基本想法:将CDR建模为一个图,Representation
K-nearest-neighbor graph;
Nodes:labeled with amino acid types;
Edges: labeled with distance & orientation。
作者提出了新的图生成方法RefineGNN,将抗体设计作为CDR生成任务,以framework region为条件。具体来说,将抗体表示一个图,该图对其序列和3D结构进行编码,并将其扩展到给定frame region为条件生成任务。最后,将RefineGNN应用于属性引导优化的抗体设计,为了简单,专注于重链CDR的生成,尽管该方法可以很容易的扩展迁移到轻链CDR的建模。
符号表示
抗体VH结构表示为氨基酸序列s=s1s2 ... sn. 每个token s_i在序列中叫残基。特殊的token<MASK>表示氨基酸类型未知,需要去预测。
抗体VH 序列会被折叠为3D结构,每个残基s_i被标记为三个骨架坐标的其一:x_i,a表示为alpha碳原子,x_i,c是它的碳原子,x_i,n是它的氮原子。
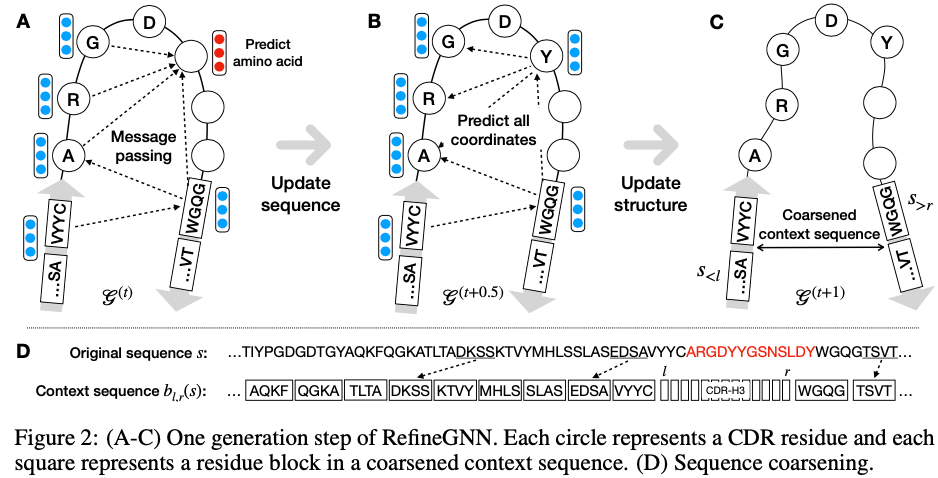
3.1 Graph Representation
每个抗体VH表示为一个图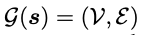
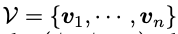
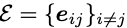
每个节点特征vi编码三个二面角(φi,ψi,ωi),与残基i的三个主干坐标相关,计算一个表示其局部坐标系的方向矩阵Oi(Ingraham et al.,2019)[3]。这允许我们计算描述两个残基i和j(即距离和方向)之间空间关系的边特征,如下所示:
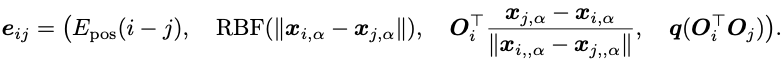
这个边特征e_ij包含四个部分:
位置编码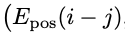
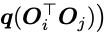
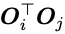
3.2 Iterative Refinement Graph Neural network(REFINEGNN)
作者建议使用迭代精调过程生成抗体图。在每一步生成步骤中,模型学习修改当前抗体图
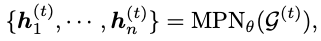
其中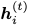
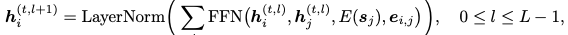
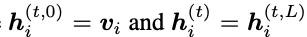
FFN是两层的前馈网络组成。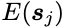
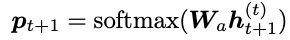
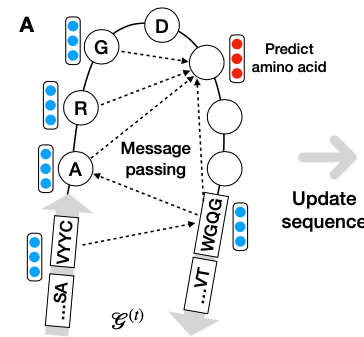
上面的预测提供了一个新的图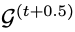
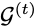
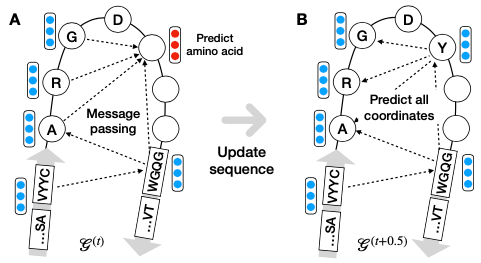
接下来需要更新结构以适应新的残基t+1。为此,用另一个不同参数θ的MPN对图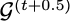
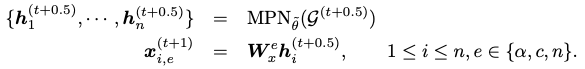
新坐标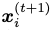
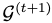
因为需要计算
结构预测(坐标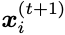
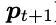
训练过程
仅将Techer forcing用于离散氨基酸类型的预测。连续结构预测在没有teacher forcing的情况下进行的。
在每次迭代中,该模型都会细化前一步预测的整个结构,并基于预测的坐标
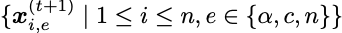
构造一个新的K-最近邻图
损失函数
模型保持旋转和平移不变,因为损失函数是在成对距离和角度而不是坐标上计算的。抗体结构预测的损失函数由三部分组成。
距离损失
对每个残基对i,j预测的alpha 碳坐标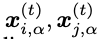
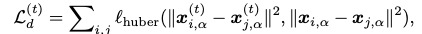
计算距离的平方,因为开根号运行有可能会导致数值不稳定。
二面角损失
对每个残基,基于预测的原子坐标
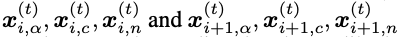
计算它的二面角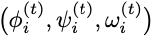
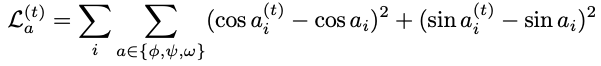
Cα角损失计算两个向量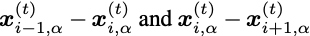
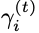
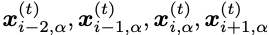
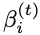
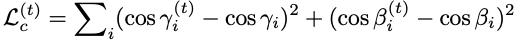
整个图生成损失定义为
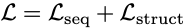
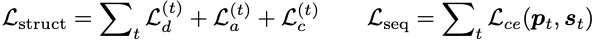
序列预测损失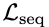
3.3 Conditional Generation Give the framework region
到目前为止,描述的模型架构师为无条件生成而设计的--它生成了一个完整的没有任何约束抗体图。
在实际中,我们通常固定抗体的框架区域(framework region),只设计CDR序列。因此,需要扩展模型结构来学习条件分布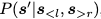
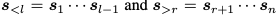

Conditioning via attention
RefineGNN的一个简单扩展是使用RNN对非CDR序列进行编码,并通过一个注意力层将信息传播到CDR。
具体的说,首先将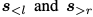

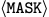

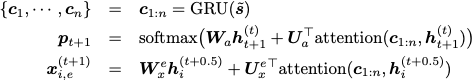
Multi-resolution modeling
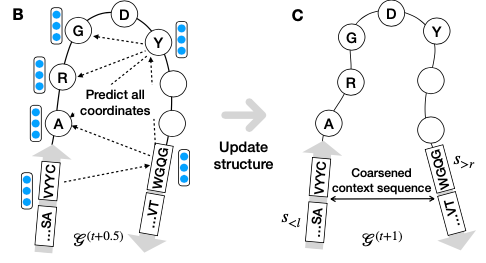
仅仅基于注意力机制是不够的,因为它没有对上下文序列的结构进行建模,因此忽略了其残基在结构上如何与CDR相互作用。虽然在测试时新抗体无法获得这些信息,但是可以学习使用已知结构的训练集中的抗体来预测这种相互作用。
一个简单的解决方案是在生成CDR残基的同时,反复细化精调整个抗体结构(超过100个残基)。但是需要重新计算每个生成步骤中所有残基的MPN编码,计算成本太高。最重要的是,我们无法一开始就预测context 残基坐标并修改它们,因为在每个生成步骤中更新CDR残基坐标时,需要在每个生成任务中重新更新。
作者提出了coarse-grained模型,将context sequence聚类成残基块来减少context sequence的长度。具体的来说,通过将每个b的context 残基聚类到一个块中来构造一个粗化的context序列

新序列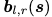
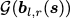
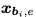
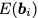
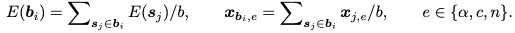
现在可以通过预测所有块的坐标,在迭代精调整个图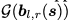
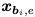

3.4 Property-Guided Sequence Optimization
终极目标是产生具有所需属性的新抗体,如中和特征病毒。这个任务可以表述为一个优化问题。设Y为是否中和的二分类变量。我们的目标是学习一个条件生成模型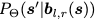
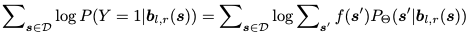
其中f(s′)是P(Y=1|s′)的预测因子。假设f给定,这个问题可以通过迭代目标增强(ITA)来解决(Yang等人,2020b)[4],在ITA优化开始之前,首先在一组真实抗体结构上预训练我们的模型,以了解CDR序列和结构的先验分布。在每个ITA微调步骤中,首先从D随机抽取一个序列s,这是一组需要重新设计CDR的抗体。接下来,根据context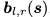
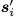
最初序列Q包含已知的中和抗体(Y=1)。最后,我们从Q中取样一批中和抗体,并通过最小化其序列预测损失
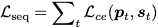
来更新模型参数。由于生成序列的结未知,因此在ITA微调阶段没有加入结构预测损失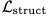
4 实验与结论
作者构建了三个评估步骤来量化该方法的性能。
首先,作者在新的抗体上衡量不同的方法的perplexity,还比较了真实的CDR与预测的结构预测误差。RefineGNN在RMSD、PPL两个指标上都显著优于所有基线模型。
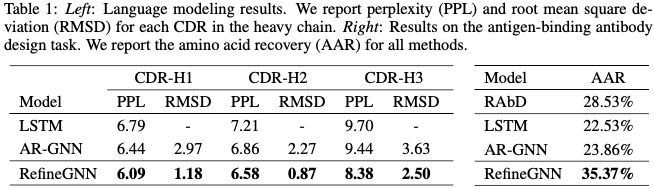
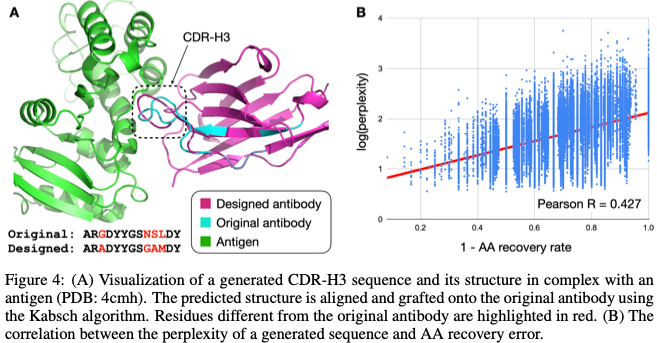
第二,在Adolf Bryfogle等人(2018)的60个抗体-抗原复合物的现有抗体设计基准上评估了该方法,目标是在给定抗原结合的情况下设计抗体的CDR-H3。ARR定义为与原始抗体中的相应残基具有相同的氨基酸的残基百分比,RefineGNN实现了最好的AAR分数,与最佳基线相比有约7%的绝对改善(Table1)。
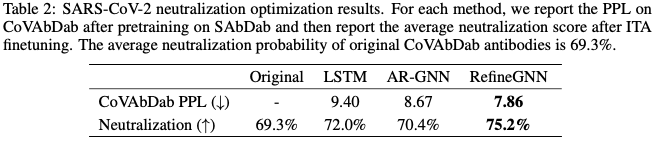
最后,提出了一项抗体优化任务,旨在重新设计冠状病毒抗体数据库中抗体的CDR-H3(Raybould等人,2021年),以提高其对SARS-CoV-2的中和作用。RefineGNN模型在平均中和score(average neutralization score)高出最佳基线3%。
参考文献
[1] Iterative refinement Graph Neural network for antibody sequence-structure Co-Design.
[2] Aditya Grover, Aaron Zweig, and Stefano Ermon. Graphite: Iterative generative modeling of graphs. In International conference on machine learning, pp. 2434–2444. PMLR, 2019
[3] John Ingraham, Vikas K Garg, Regina Barzilay, and Tommi Jaakkola. Generative models for graph- based protein design. Neural Information Processing Systems, 2019.
[4] Kevin Yang, Wengong Jin, Kyle Swanson, Regina Barzilay, and Tommi Jaakkola. Improving molec- ular design by stochastic iterative target augmentation. In International Conference on Machine Learning, pp. 10716–10726. PMLR, 2020b.
[5] Niklas WA Gebauer, Michael Gastegger, and Kristof T Schu ̈tt. Symmetry-adapted generation of 3d point sets for the targeted discovery of molecules. In Proceedings of the 33rd International Conference on Neural Information Processing Systems, pp. 7566–7578, 2019.