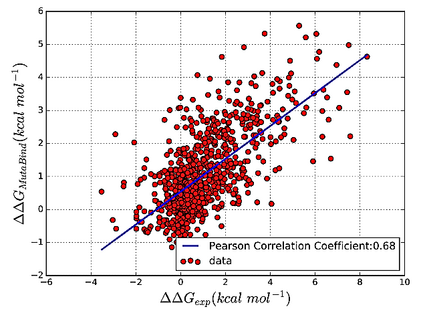
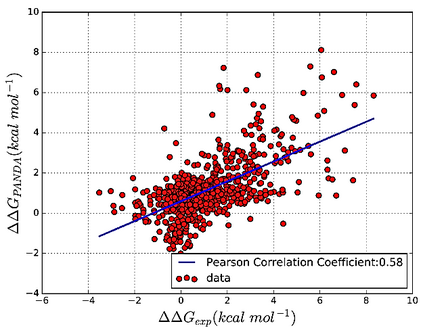
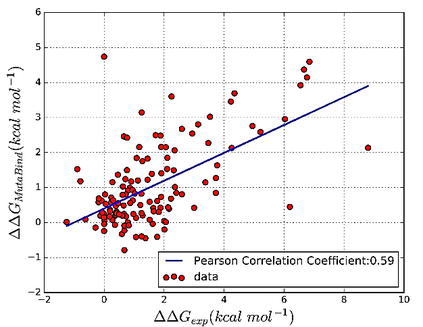
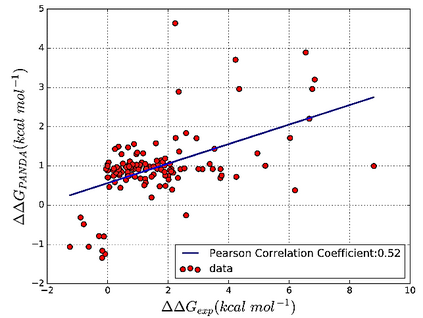
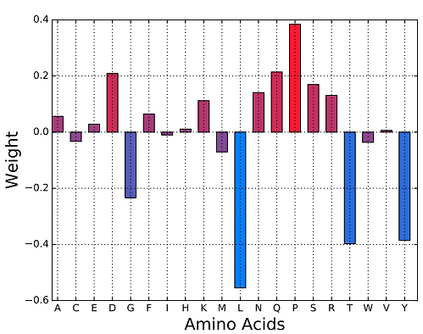
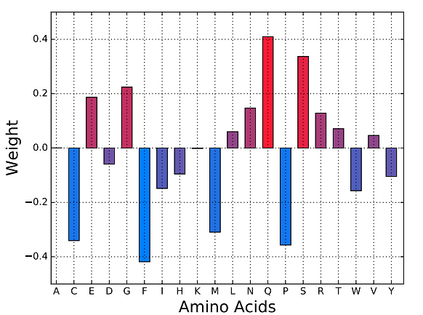
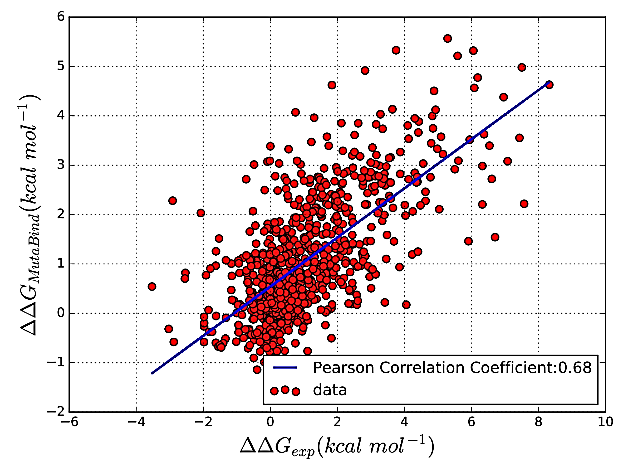
Accurately determining a change in protein binding affinity upon mutations is important for the discovery and design of novel therapeutics and to assist mutagenesis studies. Determination of change in binding affinity upon mutations requires sophisticated, expensive, and time-consuming wet-lab experiments that can be aided with computational methods. Most of the computational prediction techniques require protein structures that limit their applicability to protein complexes with known structures. In this work, we explore the sequence-based prediction of change in protein binding affinity upon mutation. We have used protein sequence information instead of protein structures along with machine learning techniques to accurately predict the change in protein binding affinity upon mutation. Our proposed sequence-based novel change in protein binding affinity predictor called PANDA gives better accuracy than existing methods over the same validation set as well as on an external independent test dataset. On an external test dataset, our proposed method gives a maximum Pearson correlation coefficient of 0.52 in comparison to the state-of-the-art existing protein structure-based method called MutaBind which gives a maximum Pearson correlation coefficient of 0.59. Our proposed protein sequence-based method, to predict a change in binding affinity upon mutations, has wide applicability and comparable performance in comparison to existing protein structure-based methods. A cloud-based webserver implementation of PANDA and its python code is available at https://sites.google.com/view/wajidarshad/software and https://github.com/wajidarshad/panda.
翻译:精确地确定突变时蛋白质结合的关联性变化对于发现和设计新型治疗方法以及协助诱变研究非常重要。 确定突变时蛋白质结合的关联性变化需要精密、昂贵和耗时的湿拉实验,这些实验可以用计算方法加以辅助。 多数计算预测技术需要蛋白结构,限制其适用于已知结构的蛋白综合体。 在这项工作中,我们探索了蛋白质结合突变时蛋白结合性变化的序列预测。 我们使用了蛋白序列信息,而不是蛋白结构以及机器学习技术,以准确预测突变时蛋白结合的关联性变化。 我们提议的蛋白质结合性新变化(称为PAAANDA)的顺序性新变化比现有的验证方法以及外部独立测试数据集的准确性更好。 在外部测试数据集中,我们拟议的方法提供了最高Pearson相关系数为0.52, 与基于州/州级现有蛋白结构的蛋白结构(称为MutaBinal-bnd)的蛋白结构, 提供了最大Pearson关联性关联性关联性系数系数系数,我们提议的蛋白质序列系统运行中的现有测试方法比0.59。 我们提出的蛋白质序列序列运行/蛋白质测试系统运行系统运行中的拟议测试法的可比较方法,对基于可比较了现有蛋白质/蛋白质/蛋白质变法的可比较方法。