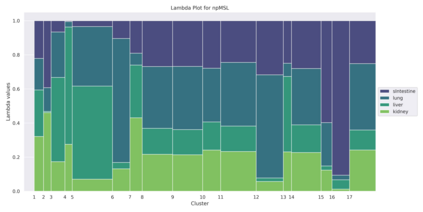
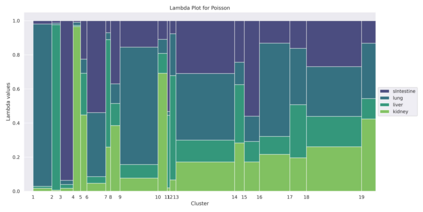
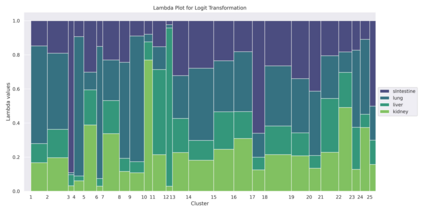
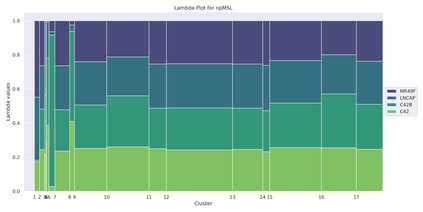
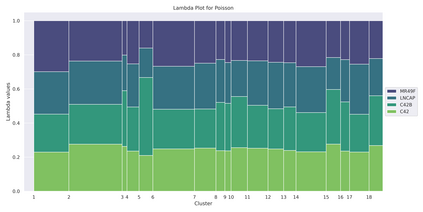
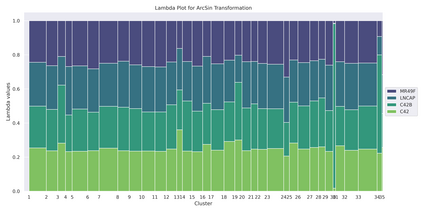
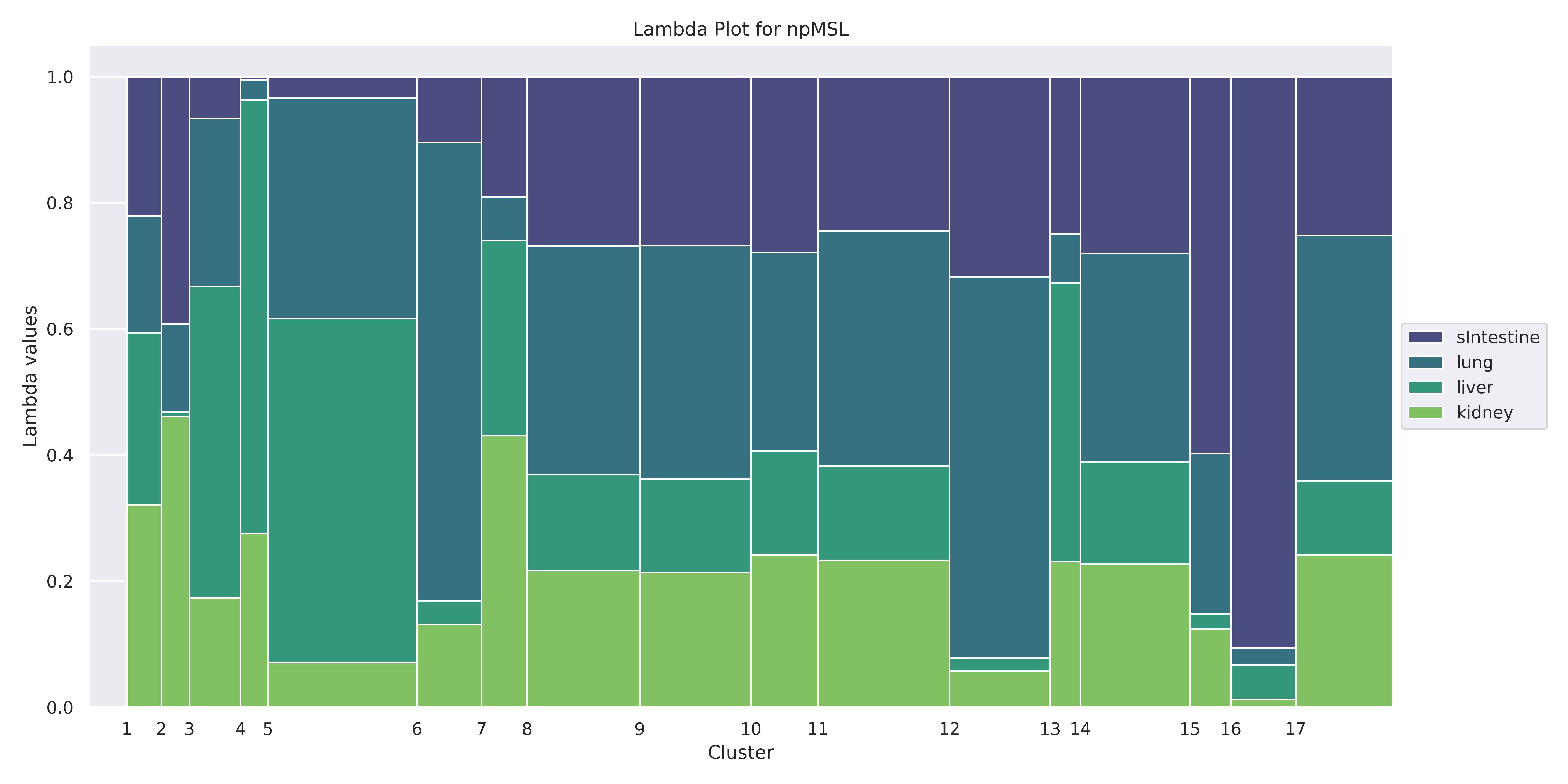
Identification of clusters of co-expressed genes in transcriptomic data is a difficult task. Most algorithms used for this purpose can be classified into two broad categories: distance-based or model-based approaches. Distance-based approaches typically utilize a distance function between pairs of data objects and group similar objects together into clusters. Model-based approaches are based on using the mixture-modeling framework. Compared to distance-based approaches, model-based approaches offer better interpretability because each cluster can be explicitly characterized in terms of the proposed model. However, these models present a particular difficulty in identifying a correct multivariate distribution that a mixture can be based upon. In this manuscript, we review some of the approaches used to select a distribution for the needed mixture model first. Then, we propose avoiding this problem altogether by using a nonparametric MSL (Maximum Smoothed Likelihood) algorithm. This algorithm was proposed earlier in statistical literature but has not been, to the best of our knowledge, applied to transcriptomics data. The salient feature of this approach is that it avoids explicit specification of distributions of individual biological samples altogether, thus making the task of a practitioner easier. When used on a real dataset, the algorithm produces a large number of biologically meaningful clusters and compares favorably to the two other mixture-based algorithms commonly used for RNA-seq data clustering. Our code is publicly available in Github at https://github.com/Matematikoi/non_parametric_clustering.
翻译:确定在笔录组群中共同表达的基因组是一项艰巨的任务。 用于此目的的大多数算法可以分为两大类: 远程法或模型法。 远程法通常使用数据对象对等的距离功能, 并将类似对象组为组群。 模型法的基础是使用混合建模框架。 与远程法相比, 模型法提供了更好的解释性, 因为每个组群都可以以拟议模型来明确定性。 然而, 这些模型在确定一种混合物可以依据的正确多变量分布方面有着特殊的难度。 在本手稿中,我们首先审查用于选择所需混合模型分布的一些方法。 然后,我们建议通过使用非参数的MSL(Mximum 平滑动的类似对象)算法来完全避免这一问题。 这种算法在统计文献中较早提出,但对于我们的知识而言,并没有应用到计算模型组的数据。 这种方法的突出特征是,它避免对单个生物样本的分布作明确的说明。 在手稿中,我们首先审查用来选择分配所需混合模型的一些方法。 然后,我们建议通过使用非参数来完全地将数据组数性地将数据组用于生物组中。