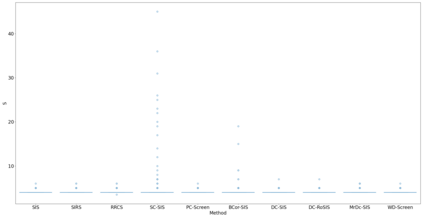
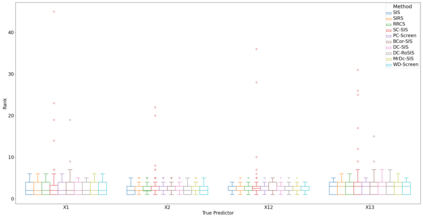
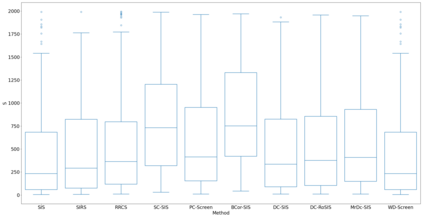
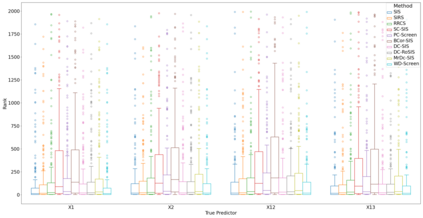
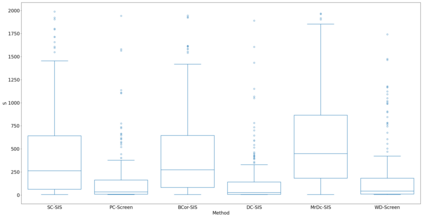
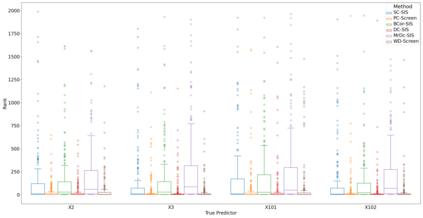
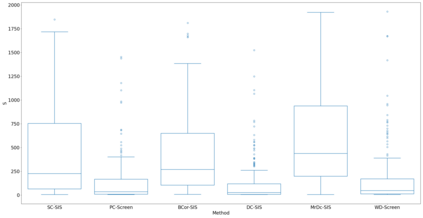
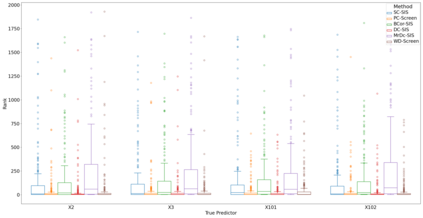
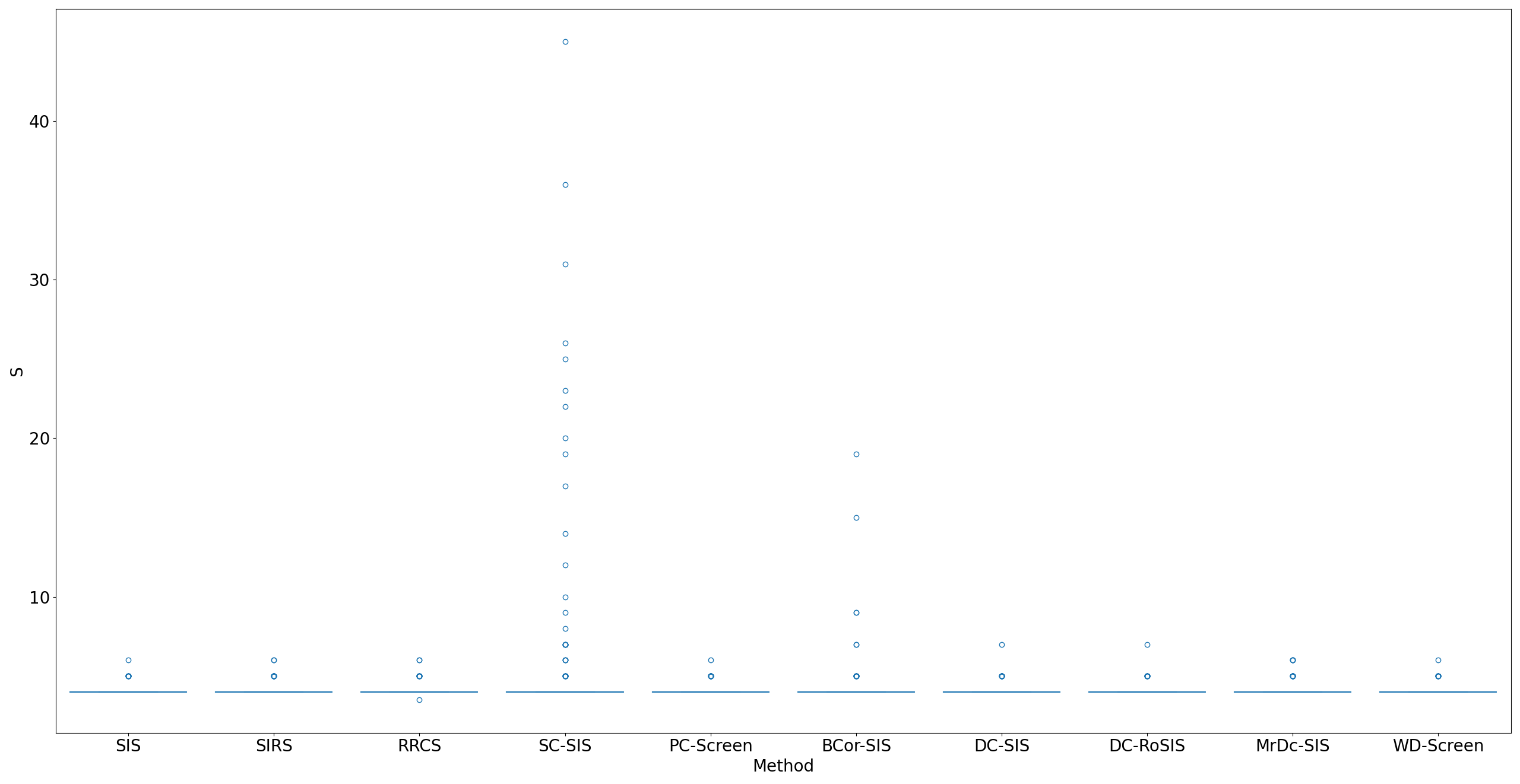
Lung adenocarcinoma (LUAD) is characterized by substantial genetic heterogeneity, posing challenges in identifying reliable biomarkers for improved diagnosis and treatment. Tumor Mutational Burden (TMB) has traditionally been regarded as a predictive biomarker, given its association with immune response and treatment efficacy. In this study, we treated TMB as a response variable to identify genes highly correlated with it, aiming to understand its genetic drivers. We conducted a thorough investigation of recent feature selection methods through extensive simulations, selecting PC-Screen, DC-SIS, and WD-Screen as top performers. These methods handle multi-omics structures effectively, and can accommodate both categorical and continuous data types at the same time for each gene. Using data from The Cancer Genome Atlas (TCGA) via cBioPortal, we combined copy number alteration (CNA), mRNA expression and DNA methylation data as multi-omics predictors and applied these methods, selecting genes consistently identified across all three methods. 13 common genes were identified, including HSD17B4, PCBD2, which show strong associations with TMB. Our multi-omics strategy and robust feature selection approach provide insights into the genetic determinants of TMB, with implications for targeted LUAD therapies.
翻译:暂无翻译