作者构建了一个蛋白质-蛋白质相互作用(PPI)靶向药物相似性数据集,并提出了一个首次用于PPI抑制剂设计的深度分子生成框架,从种子化合物的特征生成新的类药性分子。该框架使用与PPI抑制剂相关的关键特征作为输入,并为PPI抑制剂的从头分子设计开发深度分子生成模型。首次将针对PPI的抑制剂的定量类药性指标QEPPI用于PPI靶向化合物的分子生成模型的评估。结果表明生成的分子具有较好的PPI靶向药物的类药性。此外,通过化学空间分析,生成的分子与iPPI-DB抑制剂共享化学空间。
QEPPI
quantitative estimate of protein-protein interaction targeting drug-likeness https://github.com/ohuelab/QEPPI

1 背景 新药研发是一个成本高、风险大、周期长的漫长过程。新药开发涉及先导化合物的发现、优化和临床研究等多个阶段,其中早期先导化合物的发现仍是亟待解决的重要问题。传统的基于定量构效关系或基于对接的虚拟筛选通常倾向于从现有的化学库中搜索具有所需特性的分子。相比之下,从头药物设计(分子生成)旨在通过从头开始生成具有所需特性的新分子来探索类药分子的化学空间,以拓宽现有的化学库。
蛋白质-蛋白质相互作用(PPIs))在各种生物过程中发挥着至关重要的作用,因此对人类健康和疾病状态的发展至关重要。研究表明,异常 PPI 与多种疾病有关,包括癌症、传染病和神经退行性疾病。由于它们的关键影响,PPI 被认为是具有治疗意义的有希望的药物靶点。然而,以前针对 PPI 的尝试由于其一般特性而面临严峻挑战,例如平坦的表面、无特征的构象、复杂的拓扑结构和浅口袋。传统计算机引导 PPI 抑制剂的设计已取得重大进展,并且分子生成模型近年来得到了迅速发展。不幸的是,迄今为止,尚未将基于深度生成模型的方法应用于 PPI 抑制剂的设计。此外,类药性 (QED) 的定量估计通常用于评估定量药物相似性,但不适用于评估靶向 PPI 的化合物。Kosugi 和 Ohue 开发了一种针对 PPI 抑制剂的定量估计的指标 QEPPI ,专门用于评估 PPI 靶向化合物。QEPPI 是针对 PPI 靶向药物的 QED 方法的扩展,使用 QED 概念开发,涉及基于已批准药物可用信息的物理化学性质建模。研究结果表明,QEPPI 比 QED 更适合量化早期 PPI 药物发现的药物相似性。PPI 抑制剂具有两个基本的分子特征:分子形状和芳香键,因此研究人员想使用分子形状等 3D 特征作为模型的输入。
这项研究中,研究人员构建了一个 PPI 靶向药物相似性数据集,并提出了一个深度分子生成框架,以从种子化合物的特征中生成新的 PPI 靶向药物相似性分子。该研究首次探索了PPI 抑制剂分子生成模型的从头分子设计。研究人员的模型表现出与各种最先进的分子生成模型相当的性能。该模型首次将QEPPI应用于分子生成模型中生成分子的PPI药物相似性评估。结果表明,生成的分子具有更好的PPI靶向药物相似性和药物相似性。生成的分子与 iPPI-DB 共享化学空间。探索了 PPI 抑制剂的基于肽和基于配体的分子生成。
2 材料和方法 数据准备 研究人员建了一个靶向PPI的ChEMBL 的药物相似子集,用于模型训练。所有分子的拓扑数据均从 ChEMBL29 下载,并使用一系列规则清洗过滤。
模型架构 PPI 抑制剂的深度分子生成模型包括两个主要步骤:(i) GAN 使用 3D 卷积神经网络 (CNN) 来捕获分子表示,(ii) 组合CNN 和LSTM网络从分子表示中解析 SMILES。分子形状和药效团表示用作 GAN 的输入,然后是字幕网络将分子形状和药效团表示解析为 SMILES 字符串,生成与配体表示匹配的分子。
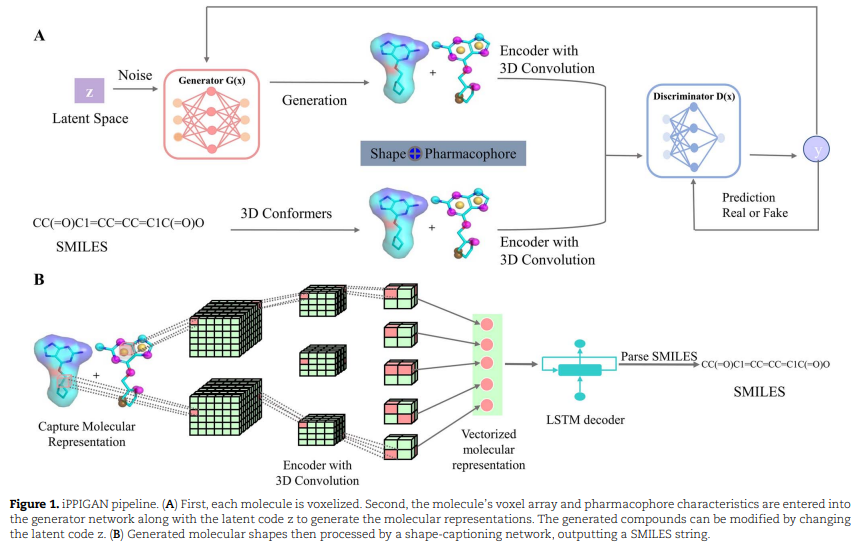
评估指标 为了深入了解模型性能并确定与其他生成模型相比的潜在优势和劣势,该方法在 MOSES 平台上进行了基准测试。
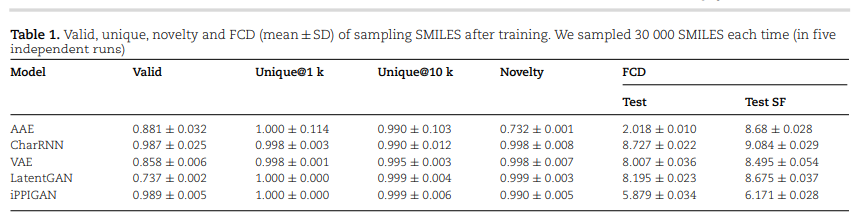
3 结果与讨论 模型性能 所有模型在来自训练数据集的 300000 个预处理的分子上进行了训练,并在 10 000 个测试和骨架拆分测试集上进行了测试。研究人员使用了 MOSES 平台中可用的模型和超参数,通过在五次独立运行中,每次采样 30000 个 SMILES 来评估该模型。
所提出的 iPPIGAN 模型优于其他具有相似或高百分比的有效、独特和新颖的 SMILES 字符串的生成模型。在 FCD 方面,研究人员开发的模型优于大多数其他方法。
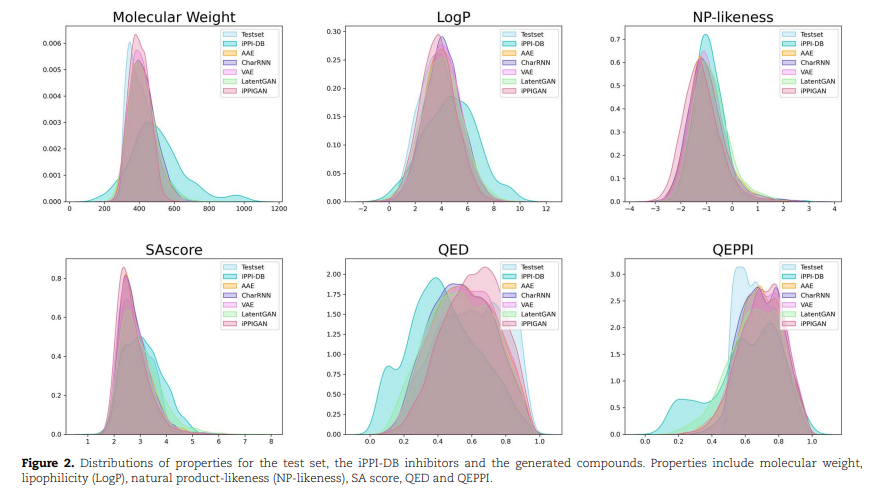
性质分布 药物设计和药物筛选领域中,LogP、合成可及性评分 SAscore、天然产物相似性 NP-likeness和 定量药物相似性QED起着基础性的作用。但 QEPPI 比 QED 更适合定量估计 PPI 靶向化合物。iPPI-DB抑制剂的QED和QEPPI平均值分别为0.43和0.61。
研究人员比较了六种分子性质的分布,即 iPPI-DB 抑制剂以及AAE、CharRNN、VAE、LatentGAN 和 iPPIGAN 生成的分子集。如图2所示,iPPIGAN生成的分子的性质分布与测试集的性质分布接近。iPPIGAN 模型生成的分子的性质分布与 iPPI-DB 抑制剂的不同,主要是因为训练数据集的性质分布与 iPPI-DB 抑制剂的不同。此外,与其他模型相比,iPPIGAN 生成的分子具有更高的 QED 值、更高的 QEPPI 值和更低的 SA 分数。结果表明,iPPIGAN生成的分子易于合成,具有更好的药物相似性和PPI靶向药物相似性。由于构建的训练集具有较高的药物相似性,该模型可以更好地学习药物相似性分布并扩展到新的药物相似性空间。
化学空间分析 为了更好地理解生成分子的化学空间,通过计算MACCS 分子指纹用作嵌入和UMAP图来评估化学空间覆盖率。如图所示,UMAP 图中,生成的分子不仅与 iPPI-DB 抑制剂共享化学空间,而且通过用新分子弥合间隙来扩展它。

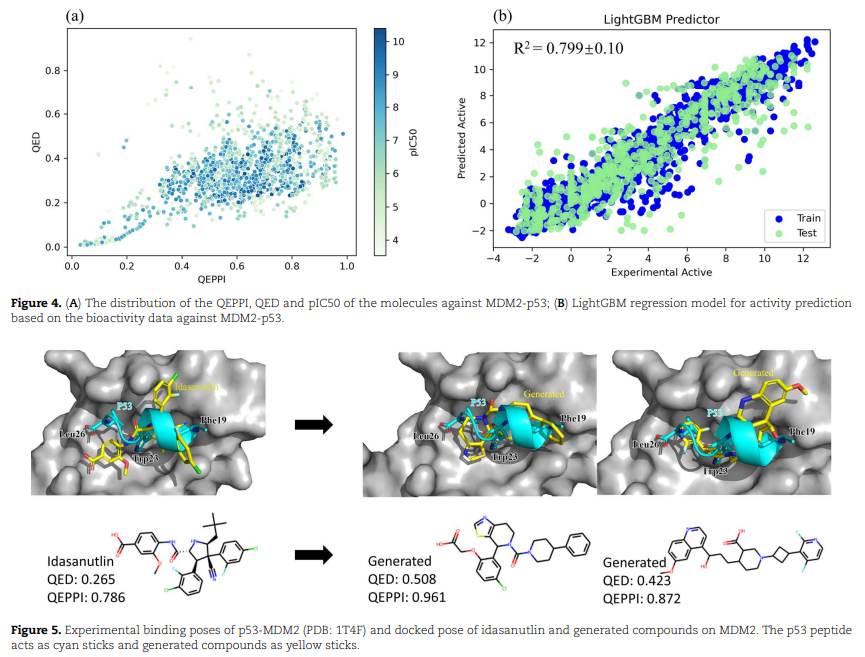
基于肽的生成分子案例 为了评估基于肽生成的化合物是否可以成为有效的候选分子,研究人员选择了基于p53(肽)生成 MDM2-p53 靶标的潜在候选抑制剂。p53 是一种有效的肿瘤抑制因子,是癌症治疗的一个有吸引力的靶标,因为它可以在功能上被激活以根除肿瘤。编码 p53 蛋白的基因在一半的人类癌症中发生突变或缺失,从而使肿瘤抑制因子的活性失活。阻断 MDM2-p53 相互作用以重新激活 p53 的功能是一种很有前景的癌症治疗策略。研究人员收集针对 MDM2-p53 的生物活性数据开发了一种用于活性预测的LightGBM回归模型,然后使用 LightGBM 回归模型来预测生成的分子是否对 MDM2-p53 PPI 靶标具有生物活性。此外,通过 QEPPI 值 > 0.5 和 SAscore值 <4 过滤化合物后,有 243 种化合物被命中。为了进一步确定优先考虑用于生物测定的分子,研究人员使用DOCK6进行分子对接来预测结合亲和力。分子对接结果显示,研究人员找到了 10 种具有比参考化合物更高的对接分数和 QEPPI 值的化合物。
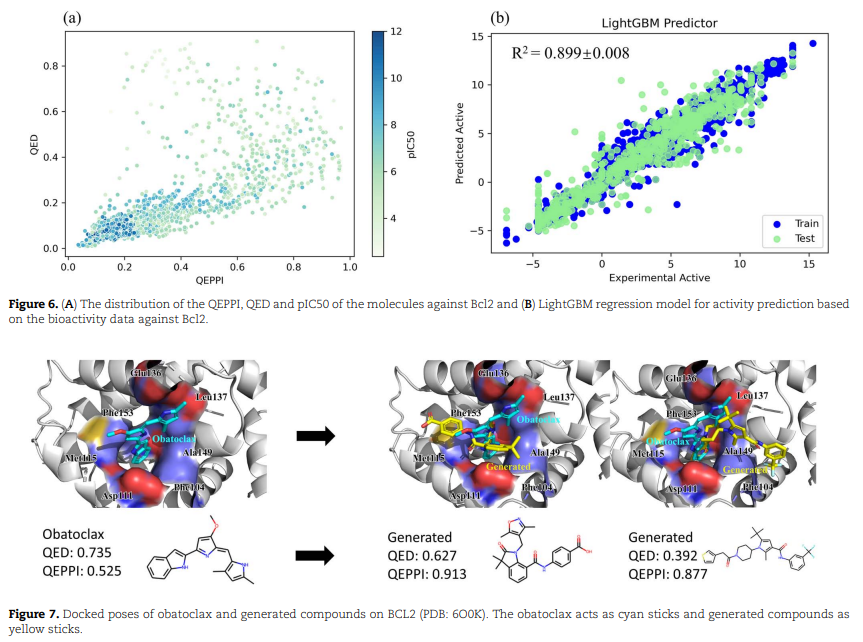
基于配体的生成分子案例 为了评估基于配体生成的化合物是否可以成为良好的候选分子,研究人员选择了基于 obatoclax 的生成 Bcl2 靶标的潜在候选抑制剂。BCL-2 蛋白家族通过控制线粒体外膜的完整性在调节细胞死亡中起关键作用。促凋亡 BCL-2 家族蛋白,如 BAK 和 BAX,在细胞凋亡中具有重要作用。当这些蛋白质被抗凋亡结合伙伴(如 BCL-2 和 BCL-XL)隔离时,它们的作用就会被阻断。通过与抗凋亡 BCL-2 家族蛋白结合来破坏这种相互作用的小分子已被设计用于诱导癌细胞的凋亡。相关的 PPI 专门针对 Bcl-2 家族蛋白,因为 Bcl-2 家族成员之间的所有相互作用都是通过 PPI。目前正在临床试验中测试三种 Bcl-2 家族 PPI 干扰物(即 navitoclax、obatoclax 和 Venetoclax)。研究人员收集针对 Bcl2 的生物活性数建立LightGBM回归模型用于针对 Bcl2 靶标的活性预测。分子指纹和分子描述符作为特征输入,IC50(nM)的自然对数作为标签,IC 50小于 100 000 nM 的值被定义为活性分子。然后使用 LightGBM 回归模型来估计生成的分子是否对 Bcl2 具有生物活性。此外,通过 QEPPI 值 > 0.5 和 SA 得分值 <4 过滤化合物后,有 44 种化合物被命中。为了进一步优先考虑用于生物测定的分子,选择性 Bcl-2 抑制剂 obatoclax 作为参考化合物,并通过对接预测结合亲和力。结果表面,其中15 种具有比参考化合物更高的对接分数和 QEPPI值。
4 结论和未来工作 这项工作中,研究人员构建了一个 PPI 靶向药物相似性数据集,并开发了一种新的基于形状的分子生成框架,用于生成针对 PPI 的新型有效药物相似性分子。研究人员的策略利用 GAN 和字幕网络从种子分子的 3D 特征生成针对 PPI 的潜在抑制剂。该方法依赖于分子或肽作为种子分子生成多样性的PPI类药性化合物库。目前是第一次将深度分子生成模型应用于 PPI 抑制剂的从头设计。此外,首次应用 QEPPI 作为分子生成模型的评估指标用于PPI靶向化合物的分子设计。研究人员的模型显示出与其他几种最先进的分子生成模型相当的性能。化学空间分析表明,生成的分子与 iPPI-DB 抑制剂具有相似的化学空间。研究人员探索了基于肽的 PPI 抑制剂设计和基于配体的 PPI 抑制剂设计。结果表明,生成的分子具有更好的 PPI 靶向药物相似性和药物相似性。PPI 在生活中无处不在,它们的研究和理解对于药物发现和生物工程的努力至关重要。PPI抑制剂的分子生成模型仍然只是向前迈出了一小步。
参考资料 Jianmin Wang, Yanyi Chu, Jiashun Mao, Hyeon-Nae Jeon, Haiyan Jin, Amir Zeb, Yuil Jang, Kwang-Hwi Cho, Tao Song, Kyoung Tai No, De novo molecular design with deep molecular generative models for PPI inhibitors, Briefings in Bioinformatics, Volume 23, Issue 4, July 2022, bbac285 https://doi.org/10.1093/bib/bbac285
数据和代码