
本文介绍一篇来自浙江大学侯廷军教授和谢昌谕教授课题组、武汉大学陈曦课题组、中南大学曹东升教授课题组和碳硅智慧联合发表的论文。该论文提出了一种全新的生成特定性质的可合成分子生成新方法ChemistGA,该算法将传统的启发式算法与深度学习算法相结合,使用基于Transformer的反应预测算法作为遗传算法杂交的核心,并且加入回交操作,不仅保留了传统遗传算法的优势,而且极大地提高了生成拥有期望特性分子的比例与可合成性。实验结果表明,ChemistGA取得了优异的表现,它为生成模型在真实药物发现场景中的应用开辟了一条新途径。

1 研究背景 新药研发是一项周期长、投入高、风险大的复杂系统工程。先导化合物的发现与优化在新药研发过程中至关重要,高质量的先导化合物能够有效缩短药物探索的时间,提高成药的可能性。在先导化合物的设计过程中,要综合考虑候选分子的生物活性、结构新颖性、靶点选择性、成药性和安全性等特性,这些性质与后续的结构修饰和优化、生物学活性评价以及临床试验密切相关。据估计,目前可开采的化学空间约为1023至1060,如何从如此庞大的化学空间中智能地发现或生成高质量的先导结构一直是从头药物设计长期努力的目标。
基于遗传算法(GA)的分子生成方法不需要模拟训练数据集的分布,因此它们表现出更强的探索能力。然而基于GA的分子生成算法存在一些固有的缺点,一是生成的分子通常难以合成,因为它们通常是通过片段的拼接替换进行生成,另一个问题是在遗传进化的过程中容易陷入局部最优。这些缺点无疑限制了GA从头设计分子的实用性。
为了解决传统基于GA的分子生成方法的缺陷,本工作将基于深度学习(DL)的正向反应预测模型与传统GA相结合,提出了ChemistGA方法及其变体Reduce-label-ChemistGA (R-ChemistGA)。ChemistGA采用的方法重新定义了GA的初始化、杂交和突变。新方法既保留了传统GA方法的优势,又大大提高了生成分子的可合成性。实验结果表明,与现有模型相比,ChemistGA和R-ChemistGA都取得了更为优秀的表现。
2 方法
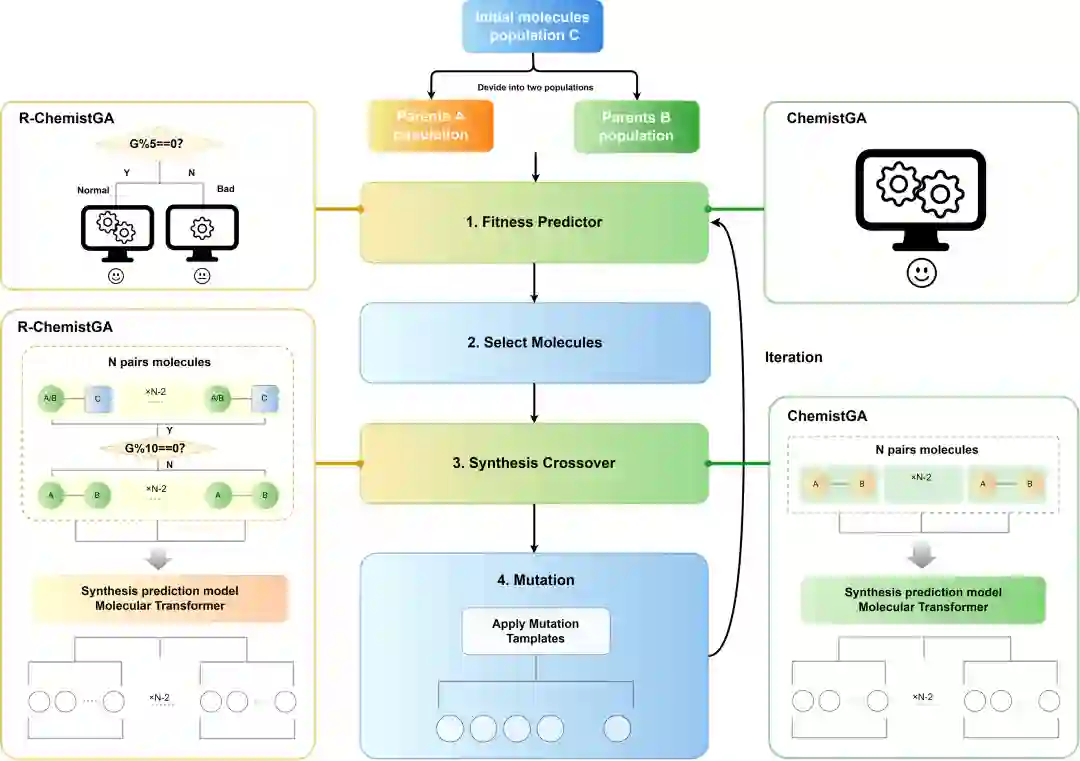
图1. ChemistGA和R-ChemistGA的工作流程,G为生成代数
ChemistGA ChemistGA算法的框架如图1所示。首先,在初始数据集C中随机选取N对分子作为初始父种子种群A和B,然后在A和B之间进行杂交,得到A和B中每对分子的推荐子代的Top-50输出;突变的第二种方法可以应用于任何后代分子,概率为1%。之后,将转换为,然后作者计算的适应度分数,并选择中分数最高的分子。最后,作者将所有的并集作为下一代分子种群池。最后含有个分子。从种群池中随机抽取下一代交叉种群和。
R-ChemistGA 在药物发现过程中,分子性质的实验验证非常耗时耗力。大多数DL生成模型尚未将这一挑战纳入设计过程。为了让ChemistGA在设计上更清楚地意识到这一限制,作者提出了一种增强的ChemistGA,简称为R-ChemistGA。图1显示了R-ChemistGA的算法流程。
使用有噪声的预测模型来估计优化分数会使计算偏离正确的路径。此外,在选择后代种群的决策中引入噪音可以提升方法的鲁棒性。最终,R-ChemistGA可以生成比原始算法ChemistGA更多样化的分子。R-ChemistGA的成功依赖于生成进化每隔几代获得正确的校准和准确的反馈。在图1所示的框架中,这种校准每隔五代就会发生一次。同时,作者在这里每隔10代进行回交,以确保一些活性结构在遗传过程中不会被消除。
实验设置 在以下场景中,作者使用靶向三个靶点的活性化合物进行评估,包括DRD2、JNK3和GSK3β。
场景1:为了显示GA的特征,将初始种群分为两组,分别命名为种群A和种群B。这两个种群分别含有靶点A和靶点B的活性分子。使用种群A和群B通过GA生成具有所需性质(靶点活性、QED,SAScore)的分子。在这个场景中,作者采用JNK3和GSK3β作为活性靶点(见任务2)。
场景2:该场景的目的是从现有的真实活性分子中生成新的期望分子(初始分子是真正的期望分子)。为了使结果更有说服力,作者考虑了与真实世界药物发现相关的两项任务,并设计了针对这些靶点的活性靶点和理化性质的组合。 * 任务1:DRD2、QED和SA。
在这项任务中,目标是生成DRD2活性≥0.5、QED≥0.6和SA≤4的分子。
任务2:JNK3、GSK3β、QED和SA。
在这项任务中,目标是生成JNK3、GSK3β活性≥0.5、QED≥0.6和SA≤4的分子。
这里所生成的具有所需性质的分子被称为“成功分子”,可以被合成的“成功分子”被称为“完美分子”。“成功分子”与总生成分子的比率被定义为成功率。
杂交 ChemistGA中的杂交过程是通过正向反应预测模型Molecular Transformer(MT)实现的。该模型以一对分子作为输入(格式为SMILES.SMILES),并输出转换后的SMILES。但作者指出不应严格按照反应的角度理解这个杂交过程,作者将在化学合成预测部分进一步解释这一点。当在GA工作流中使用MT时,MT被输入任意分子对,这些分子对是极有可能不会发生化学反应的,然而,MT依然会推荐新的分子。为了在广阔的化学空间中引导分子种群进化走向合适的区域,从MT每次预测两个输入分子给出的前50个分子中评分最高的3个分子,这3个分子将被添加到后代种群池中。每次选择3个后代也可以让生成的结果保持更多样化的后代群体。
变异 在此框架下,突变以两种形式出现。一个是MT推荐的随机性,更准确地说,生成模型MT并没有严格地按照一套简单的规则杂交两个输入分子。许多输出分子不仅继承部分父代结构,而且可能插入全新的片段。这是因为MT是用反应数据集训练的,许多反应会添加、替代或移除分子片段(反应预测的误差)。
第二种突变类型与Virshu等人使用的突变类型相似,作者将随机将SMART定义的反应模板应用于分子,以产生模板定义的突变。这些突变可分为7大类,具体如下:1、附加原子(15%);2、插入原子(15%);3、删除原子(14%);4、改变原子类型(14%);5、改变键的顺序(14%);6、删除环中的键(14%);7、在环中添加键(14%)。
优化函数 **标准优化函数:**SA评分为二值函数,满足时为1,不满足时为0。QED和DRD2或其他生物活性评分是0到1之间的连续值。

**离散优化函数:**所有分数均采用二进制表示设计,满足要求时为1,不满足要求时为0。

活性预测模型 作者训练了两种类型的预测模型:一种是标准的预测模型,另一种是精度明显较低的预测模型。所有模型均在DRD2、JNK3和GSK3β数据集上训练,以摩根分子指纹作为输入,随机森林分类模型作为标准预测模型,最后以分类模型预测的阳性概率作为生物活性评分。
基准模型 本研究以RationaleRL、REINVENT和GB-GA这3个在多约束分子设计任务中表现出色的模型作为基线。
一个经过训练的RationaleRL模型被用作任务2的基线。因为RationaleRL需要大量的时间来从头开始训练,所以作者没有在任务1中包含RationaleRL的结果。REINVENT在他们报道的数据集中进行了基于强化学习的预训练(该数据集包含来自ChEMBL的超过100万个分子),以便在任务中进行微调。
评价标准 论文中采用的评价标准分别为完美分子可合成率、多样性、新颖性、骨架数量,其中完美分子可合成率由Retro*逆合成规划算法预测得到;多样性是基于Tanimoto距离计算得到;新颖性的标准被定义为生成的分子与其最近邻居之间的相似性低于0.4;分子骨架由Murcko骨架提取算法计算。
3 结果与讨论 场景1 场景1实验是证明ChemistGA优于其他传统GA算法,如GB-GA。作者的基准指标包括比较每代分子的最高分数,以及每代中成功分子与所有生成分子的比率。图2左侧第一行显示了两种方法(ChemistGA和GB-GA)生成分子的成功率与生成代数的关系,其中ChemistGA的成功率随着进化代数的增加而持续上升,最终稳定在0.45左右。然而,在整个进化过程中,GB-GA的成功率始终保持在0左右,这意味着GB-GA不能结合两种分子的优势成功地产生后代。
然后,作者通过拆分四个评分(图2的剩余子图)来比较GB-GA和ChemistGA。在进化过程中,ChemistGA在除SA(开始时已经很高)之外的所有评分上都显示出明显的群体优化趋势,而GB-GA的群体优化对于JNK3评分来说极其缓慢甚至退化。GB-GA很有可能是随机切断亲本分子,然后随机组合产生子代分子,从而产生具有高度随机性(分子结构差异巨大)的子代群体,由于消除了大量分子,非常不利于活性结构的遗传,使得优化效率极低。
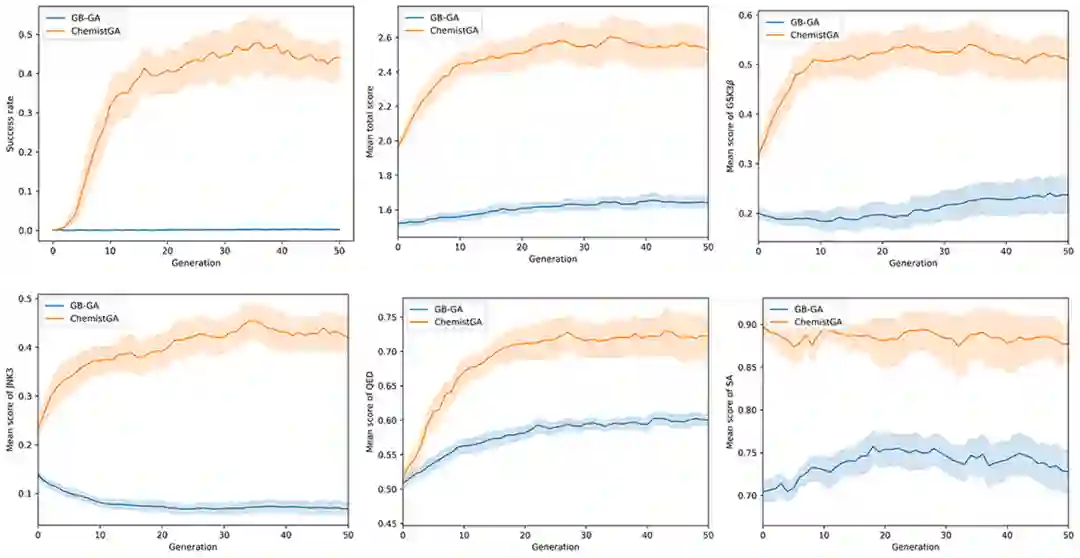
图2. 在场景1中分析两种方法的分子性质和生成之间的关系。
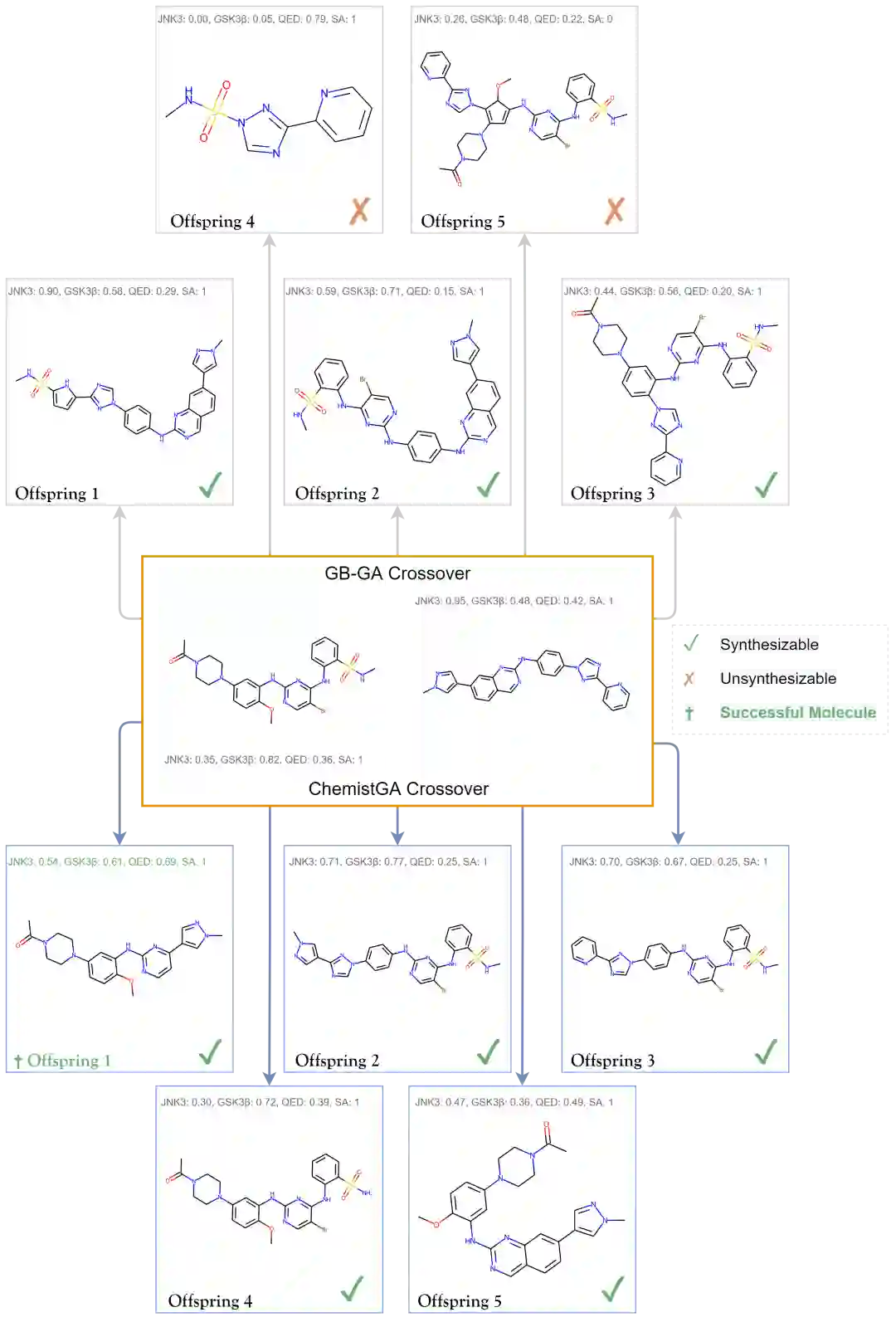
图3. ChemistGA和GB-GA的单步杂交结果示意图。
如图3所示,ChemistGA与GB-GA相比,采用了化学反应约束的杂交方法,对分子结构的继承更加合理,对四个任务的优化效率更高,使所生成的后代更有可能保留亲代的子代结构,并确保了后代的可合成性。相比之下,GB-GA在杂交过程中采用随机剪切和拼接策略(50%环内剪切和50%非环内剪切,随机确定切割位点)。这种杂交方法容易破坏活性分子的结构,不能保证生成分子的可合成性。而且,随着代数的增加,低分数的后代(即GB-GA中的分子4和5)可能会聚集并在新生成的后代分子中占优势,使得高分数的分子所占的比例逐渐变小,逐渐从生成的分子集合中消失。
场景2 作者在这个场景中用多个指标来评估生成的分子,包括分子合成率、多样性、新颖性和包含的分子骨架数量,随机选择5000个具有期望性质的生成分子来评估每个模型。作者将生成的可以合成的分子称为完美分子,并对其新颖性、多样性和骨架进行了评估。
在任务1中,对REINVENT、GB-GA和ChemistGA进行了基准测试;在任务2中,对REINVENT 、RationaleRL、GB-GA和ChemistGA进行了基准测试。正如前面所解释的,因为RationaleRL的训练需要太高的计算成本,并且RationaleRL的原始工作的实验设置只与任务2匹配,所以作者在只在任务2中单独应用RationaleRL。
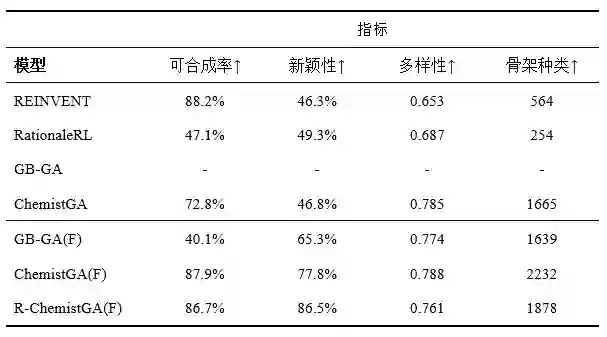
任务1结果 如表1的上半部分所示,虽然在可合成性方面,REINVENT取得了最好的性能,但它的新颖性出乎意料地低,这意味着生成的分子与真正的活性化合物非常相似。此外,在三种测试方法中,它提供的独特骨架的多样性和数量最少。对于GB-GA,可合成性和新颖性都很低。因此,这两种方法都无法与ChemistGA竞争,而ChemistGA在这些评估指标上实现了更全面的性能。此外,与REINVENT相比,GB-GA和ChemistGA在分子新颖性、多样性和骨架种类方面具有压倒性的优势。
表1. 比较场景2-任务1中各种算法生成的分子区别。
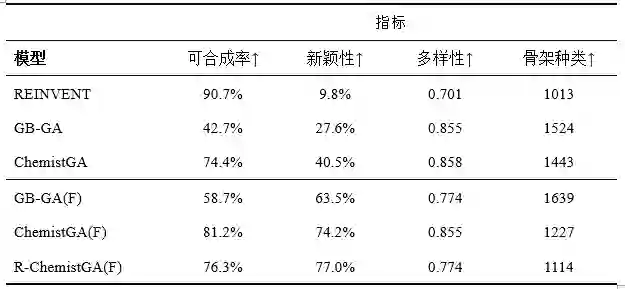
在任务2中的表现 表2的上半部分显示了任务2的性能。在这项任务中,GB-GA在成功合成分子方面面临着重大挑战。对于REINVENT,尽管它取得了令人印象深刻的88.2%的合成率,但在分子多样性和新颖性方面表现不佳。RationaleRL的合成率为47.1%,ChemistGA的合成率为72.8%。在新颖性方面,三种模型(除GB-GA外)表现出比较接近的结果,新颖性得分在45%~50%之间。在多样性方面,REINVENT和RationaleRL的表现都比ChemistGA差(多样性分数分别为0.653和0.687),ChemistGA的多样性分数为0.785。唯一骨架的数量差距最大,其中ChemistGA生成的骨架(完美分子)数量为1665个,明显高于REINVENT和RationaleRL生成的骨架数量(分别为564和254个),这意味着ChemistGA的骨架数量远高于纯机器学习生成模型(即REINVENT和RationaleRL)生成的骨架数量。此外,与GB-GA等传统遗传算法相比,ChemistGA在生成具有良好QED性质和可合成性的成功分子方面表现出了更优越的能力。
表2. 比较场景2-任务2中各种算法生成的分子区别。
使用离散优化策略性能 对于场景2中的两个任务,ChemistGA表现出更平衡的性能,在所有评估指标上没有明显的缺陷。虽然ChemistGA作为一种基于GA的算法在可合成性方面已经做得很好(参见表1中的GB-GA的数值),但作者预计它的可合成性可以被进一步提高,以缩小它与REINVENT之间的差距。为了解决这个问题,作者决定对所有基于GA的算法使用离散适应度函数,并重新评估它们在相同任务中的表现。
如表1和表2的下半部分所示,使用离散适应度评分后,GB-GA和ChemistGA的新得分得到了很大的提高,其中ChemistGA和GB-GA在任务1中的新得分分别提高了33.7%和35.9%,而ChemistGA在任务2中的新新得分提高了31.0%。
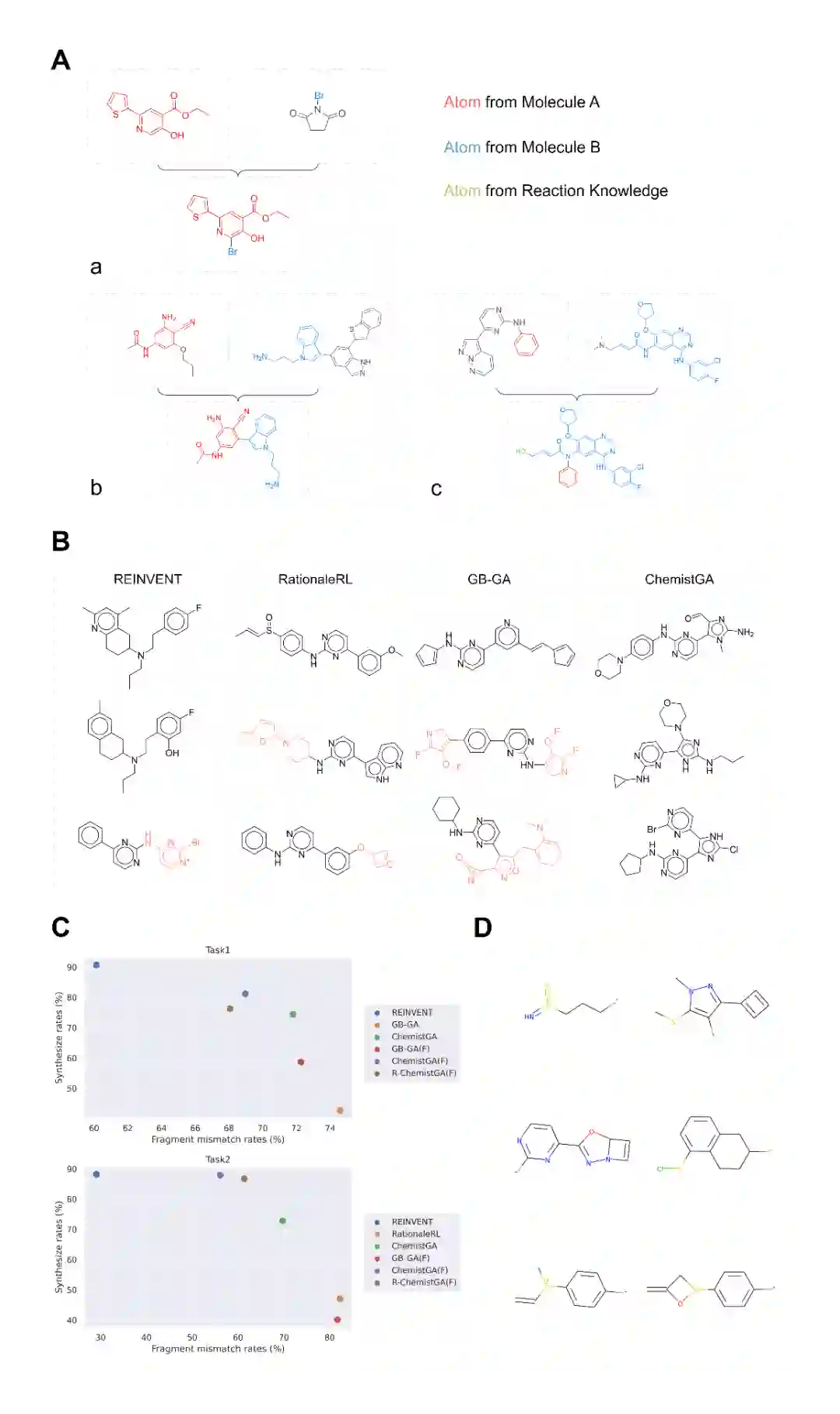
图4. 生成分子的可合成性解释。
为了更直观地反映生成的分子,作者展示了一个样例,从图4的B图中可以看出,由REINVENT生成的分子更合理,但分子相似、结构单一,RationaleRL和GB-GA生成的分子更多样,但存在一些明显的错误。其中分子的红色部分是Retro*算法找不到任何逆合成模板的子结构。ChemistGA合成的分子多样性高、合成率高,未发现明显的不合理分子,与以上报道的评价指标高度一致。
为了进一步证明MT作为杂交核心的作用,作者在图4 A中总结了三种交叉行为(这里A和B代表输入分子,C代表输出分子):(1)输入 输出正常反应产物;(2)输入 输出A/B子结构组装的;(3)输入 输出A/B子结构+MT化学反应知识库中所包含的官能团组装的 。MT中的杂交包含了杂交和突变操作。最后,作者在图4D中可视化了一些与可购买的分子数据集的片段不匹配的分子片段,并发现这些分子片段大多数包含化学上不稳定的基团。这一结果也证实,所生成的分子中含有的化学不稳定基团的百分比越高,它们的合成能力就越低。
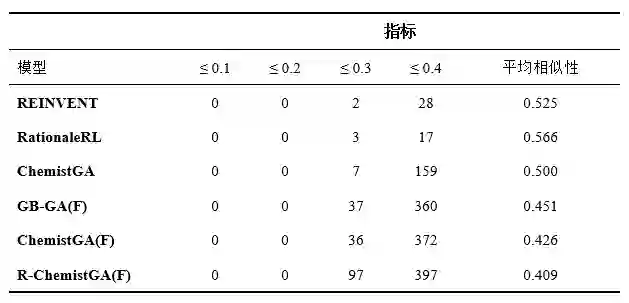
骨架分析 在本节中,作者对任务2中的1,000个分子进行了Murcko骨架分析,将其与真正的活性化合物的骨架进行比较。记录生成的分子骨架与活性化合物的骨架之间最高的相似性。表3显示了相似度小于或等于某一数值的分子骨架数量。最后一行是相似度的平均值。从表3可以看出,GA模型在骨架新颖性方面比REINVENT和RationaleRL表现得好得多。ChemistGA和ChemistGA(F)的比较表明,离散的优化策略可以使GA在一定程度上跳出局部最优,从而生成更多的新型骨架。
表3. 真实活性分子和生成分子之间骨架的相似性。
4 总结 综上所述,作者提出了两种分子生成算法ChemistGA和R-ChemistGA,它们在GA框架下加入了Molecular Transformer来驱动基于反应的进化策略。与纯机器学习的生成模型相比,这些算法具有以下优点:无需训练模型,所需的初始数据较少,生成的分子结构多样性较高。此外,与基于遗传算法的生成模型相比,生成的分子结构具有更高的可合成性和多样性。除了突出的可合成性,ChemistGA在许多其他指标上也表现出非常令人印象深刻的表现,如新颖性、多样性和骨架。与基准中的其他模型相比,ChemistGA在几乎所有的评价指标下都具有明显的优势。此外,为了解决真实药物发现过程中难以标记分子性质的问题,作者还提出了一种鲁棒性更高的算法R-ChemistGA。具有ChemistGA的所有优点,R-ChemistGA还大大减少了对准确预测模型的依赖,当使用相同数量的准确预测模型时,可以生成比ChemistGA多两倍以上的所需分子,这证明了R-ChemistGA对实际应用中经常出现的标签的灵活性。使模型更适合用于真实世界的药物从头设计。虽然目前将分子生成应用于真正的药物发现还存在许多困难,但是ChemistGA的出现可能为药物从头设计提供一些启示。 参考资料 ChemistGA:A Chemical Synthesizable Accessible Molecular Generation Algorithm for Real-World Drug Discovery, Journal of Medicinal Chemistry, 2022. https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01179
