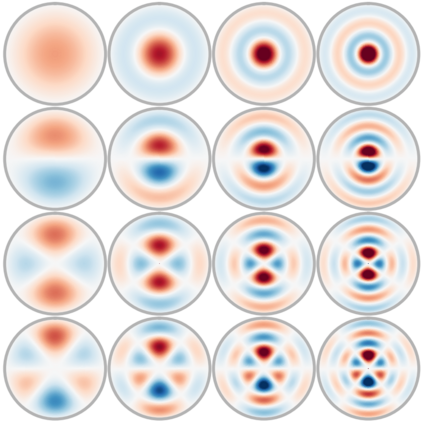
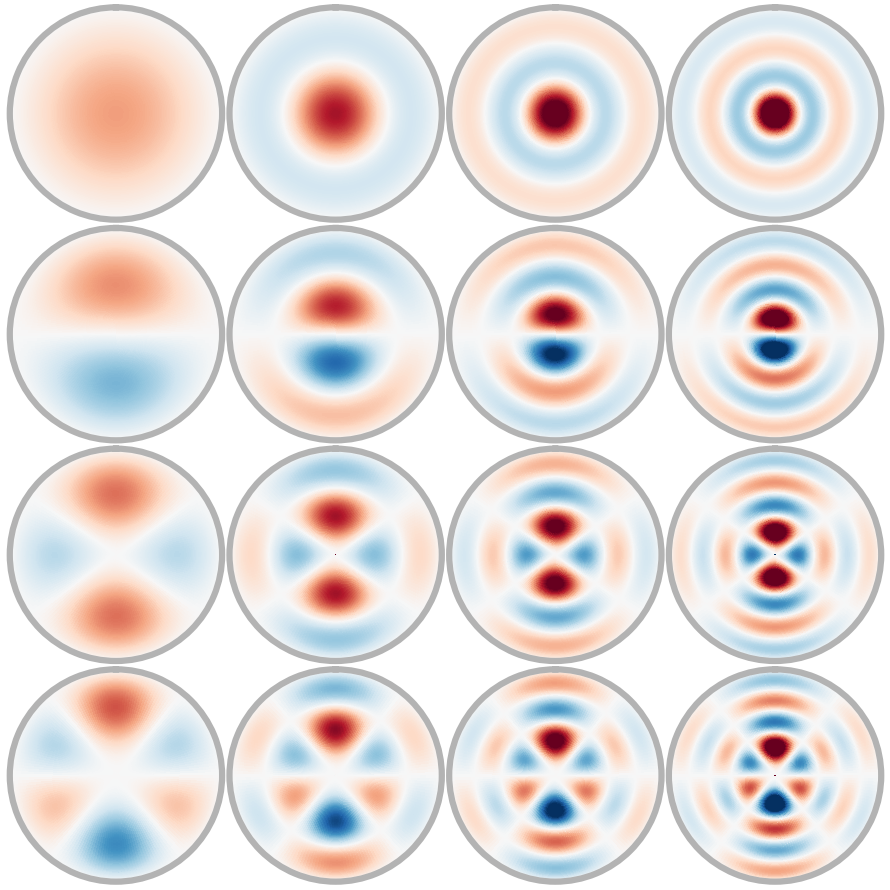
Effectively predicting molecular interactions has the potential to accelerate molecular dynamics by multiple orders of magnitude and thus revolutionize chemical simulations. Graph neural networks (GNNs) have recently shown great successes for this task, overtaking classical methods based on fixed molecular kernels. However, they still appear very limited from a theoretical perspective, since regular GNNs cannot distinguish certain types of graphs. In this work we close this gap between theory and practice. We show that GNNs with directed edge embeddings and two-hop message passing are indeed universal approximators for predictions that are invariant to translation, and equivariant to permutation and rotation. We then leverage these insights and multiple structural improvements to propose the geometric message passing neural network (GemNet). We demonstrate the benefits of the proposed changes in multiple ablation studies. GemNet outperforms previous models on the COLL, MD17, and OC20 datasets by 34%, 41%, and 20%, respectively, and performs especially well on the most challenging molecules. Our implementation is available online.
翻译:有效预测分子相互作用有可能通过多个数量级加速分子动态,从而实现化学模拟的革命性。 图形神经网络(GNNS)最近为这项任务展示了巨大的成功,超过基于固定分子内核的古典方法。 但是,从理论角度看,这些网络似乎仍然非常有限,因为普通的GNNS无法区分某些类型的图表。 在这项工作中,我们缩小了理论和实践之间的差距。 我们显示,带有定向边缘嵌入和双跳信息传递的GNNs确实与无法翻译的预测和不可变换和旋转的等同体。 然后,我们利用这些洞见和多重结构改进来提出几何信息传递神经网络(GemNet) 。 我们展示了在多动关系研究中拟议变化的好处。 GemNet将COLL、MD17和OC20数据集的先前模型分别比34%、41%和20%的模型更完美,并在最具挑战的分子上表现得特别好。