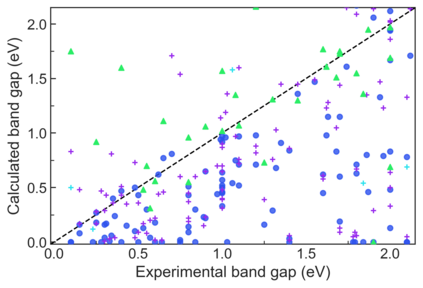
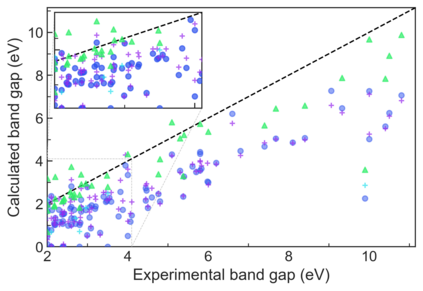
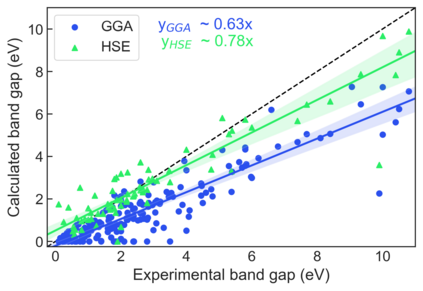
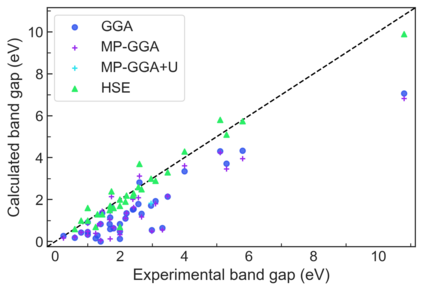
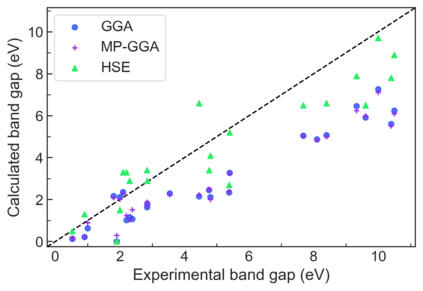
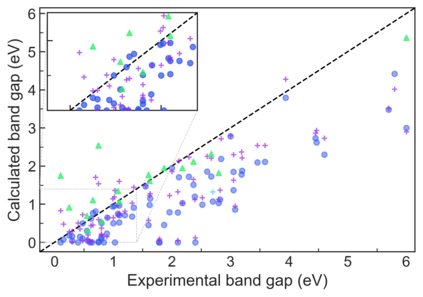
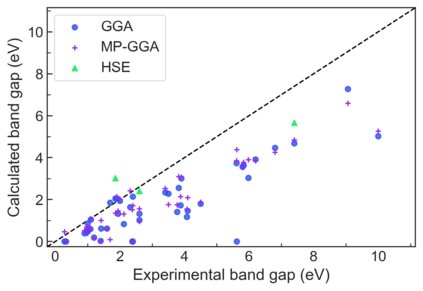
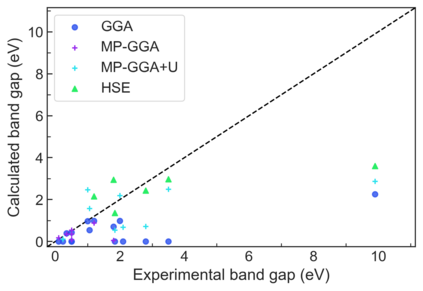
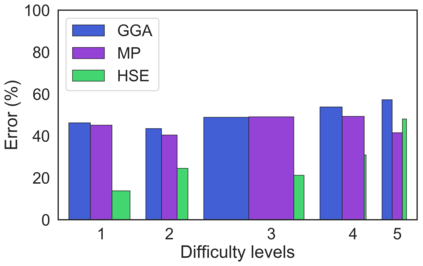
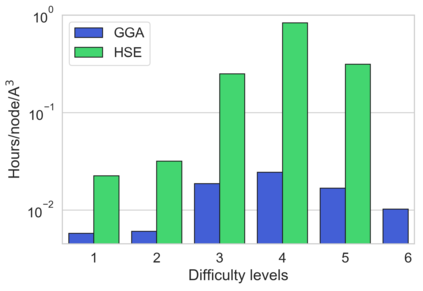
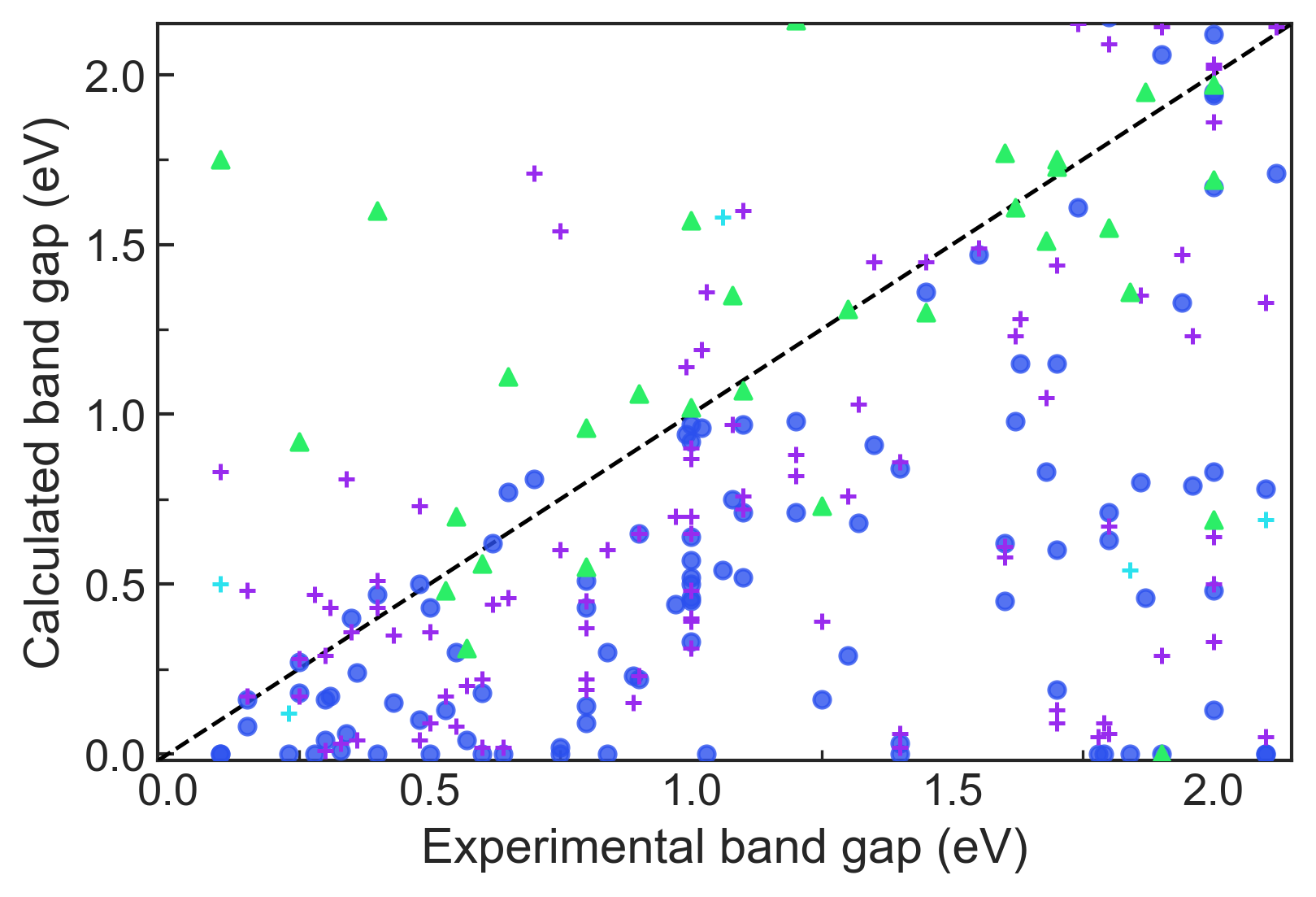
We present example applications of an approach to high-throughput first-principles calculations of the electronic properties of materials implemented within the Exabyte.io platform. We deploy computational techniques based on the Density Functional Theory with both Generalized Gradient Approximation (GGA) and Hybrid Screened Exchange (HSE) in order to extract the electronic band gaps and band structures for a set of 775 binary compounds. We find that for HSE, the average relative error fits within 22%, whereas for GGA it is 49%. We find the average calculation time on an up-to-date server centrally available from a public cloud provider to fit within 1.2 and 36 hours for GGA and HSE, respectively. The results and the associated data, including the materials and simulation workflows, are standardized and made available online in an accessible, repeatable and extensible setting.
翻译:我们举例说明了对Exabyte.io平台内实施的材料电子特性电子特性的高通量第一原则计算方法的应用。我们采用基于密度功能理论的计算技术,同时采用通用逐步接近和混合屏幕化交换(HSE),以便为775种二元化合物抽取电子带间隙和带状结构。我们发现,对于HSE来说,平均相对误差在22%之内,而对于GGA来说是49%。我们发现,从公共云源供应商中央可得到的最新服务器上的平均计算时间分别符合GGGA和HSE的1.2小时和36小时。结果和相关数据,包括材料和模拟工作流程,都标准化,在可访问、可重复和可扩展的环境中在线提供。