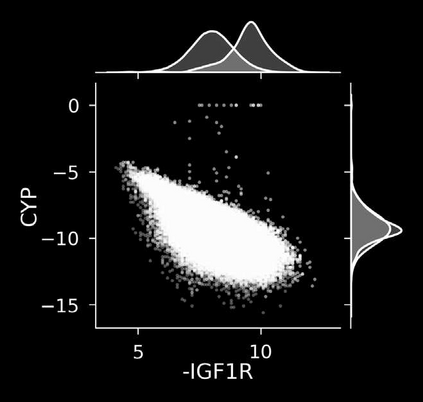
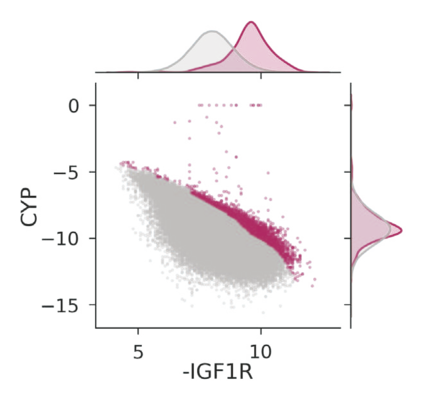
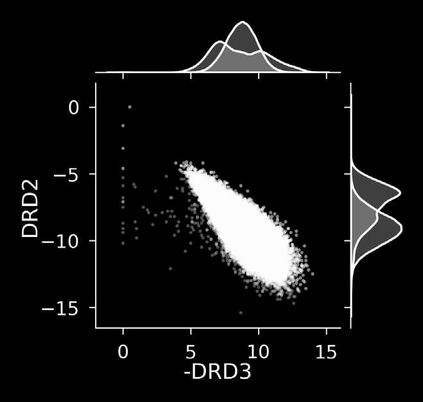
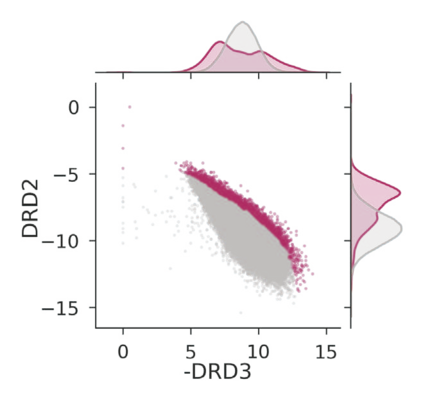
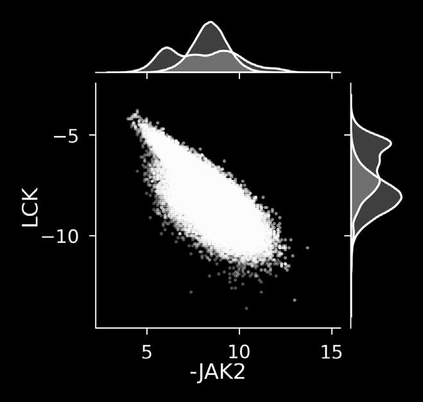
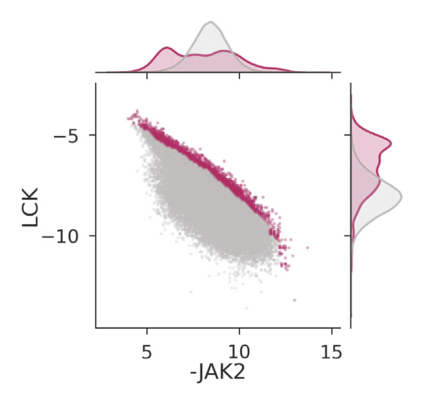
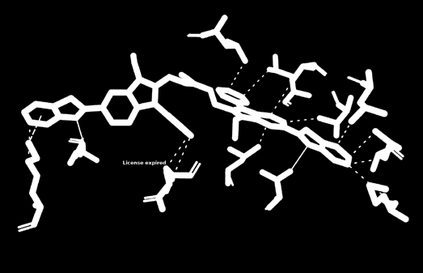
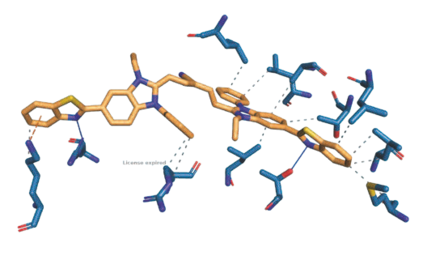
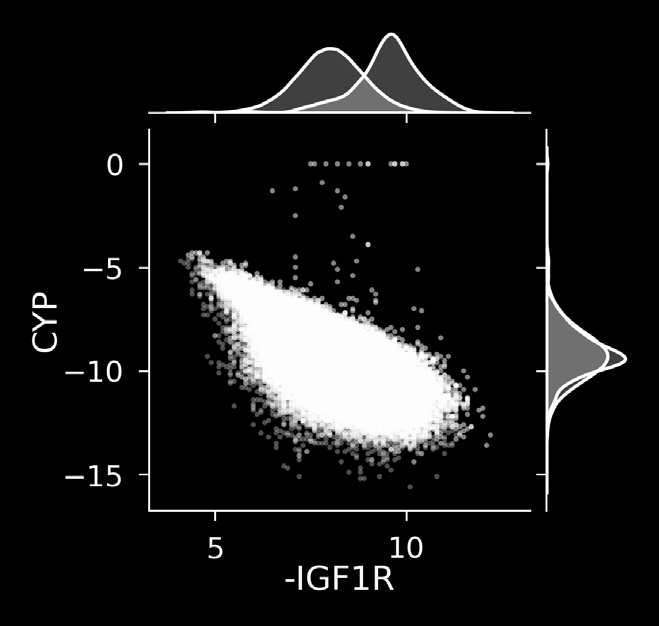
The discovery of therapeutic molecules is fundamentally a multi-objective optimization problem. One formulation of the problem is to identify molecules that simultaneously exhibit strong binding affinity for a target protein, minimal off-target interactions, and suitable pharmacokinetic properties. Inspired by prior work that uses active learning to accelerate the identification of strong binders, we implement multi-objective Bayesian optimization to reduce the computational cost of multi-property virtual screening and apply it to the identification of ligands predicted to be selective based on docking scores to on- and off-targets. We demonstrate the superiority of Pareto optimization over scalarization across three case studies. Further, we use the developed optimization tool to search a virtual library of over 4M molecules for those predicted to be selective dual inhibitors of EGFR and IGF1R, acquiring 100% of the molecules that form the library's Pareto front after exploring only 8% of the library. This workflow and associated open source software can reduce the screening burden of molecular design projects and is complementary to research aiming to improve the accuracy of binding predictions and other molecular properties.
翻译:暂无翻译