两篇JCM文章|机器学习对DEL苗头化合物发现建模
近日,JCM上发表了题为“Machine Learning on DNA-Encoded Library Count Data Using an Uncertainty-Aware Probabilistic Loss Function“的文章。作者展示了一种回归方法来学习单个分子的DEL富集,使用了自定义的负对数似然损失函数,该函数有效地对DEL数据进行了去噪,并为学习的结构-活性关系的引入了可视化方法。

DNA编码库(DEL)筛选和定量构效关系(QSAR)建模是药物发现中用于寻找结合蛋白质靶标的新型小分子的两种技术。将QSAR模型应用于DEL挑选数据有助于用于化合物的off-DNA合成和评估。目前这种组合方法通过训练二元分类模型无法区分不同富集级别,并且在“disynthons”(所有分子具有两个共同的构建块)聚合过程中可能会丢失信息。
作者对DEL实验工作流程中测序过程的泊松统计进行了建模。在针对碳酸酐酶 (CAIX) 筛选的 108,528 种化合物的 DEL 数据集和针对可溶性环氧化物水解酶 (sEH) 和 SIRT2 筛选的 5,655,000 种化合物的数据集上说明了这种方法。
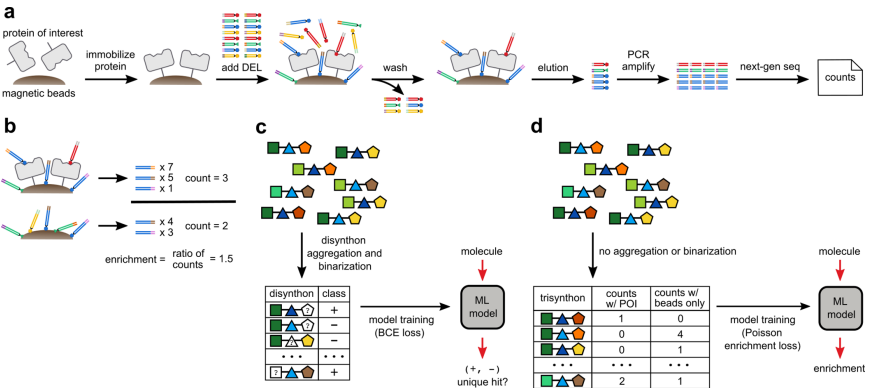
分析DNA编码文库(DEL)选择数据以推断结构-活性的方法。(a)DEL实验工作流程(b)获取hits的传统方法:原始归一化计数(c)McCloskey [1]等人的方法:“disynthons”的二分类方法(d)作者的方法:对“trisynthons"的回归任务,考虑到不确定性。
虽然 QSAR 建模在小分子药物发现中无处不在,但直到最近McCloskey等人的方法[1]才应用于 DEL。通过对分子进行分组并使用它们的聚合富集数据将每个组标记为 POI 的竞争性粘合剂,将 QSAR 建模应用于 DEL 数据——一个二元分类问题(图1c)。在 disynthons 级别聚合分子部分减轻了 DEL 数据的稀疏性并增加了分配标签的确定性。
QSAR 模型接受了好/坏disynthons的训练,并用于在虚拟筛选库中筛选化合物,最终成功识别出具有微摩尔活性的先导化合物。
然而,这种建模方法的两个潜在缺点是(a)模型无法区分不同的富集水平(例如,1.5 和 10.0 的值都是二进制意义上的“富集”,但不是同样如此)和(b)关于单个分子的富集在聚集过程中丢失。
作者探索了一种在回归公式中结合 QSAR 建模和 DEL 数据的替代方法,该方法避免了富集数据的二值化和聚合(图1d)。虽然作者的方法没有像 McCloskey等人[1](即下面那篇文章)那样展示推广到新化学结构的能力。但确实表明,由于在训练期间使用了新颖的不确定性感知损失函数,模型可以有效地对 DEL 数据进行去噪。
作者使用了概率损失函数,认为它比标准回归损失函数(如均方误差 (MSE))更适合建模 DEL 数据,因为数据获取方式具有随机性。因为 DEL 合成的二代测序步骤很好地符合泊松分布,作者定义一个损失函数来解释泊松采样导致的不确定性。在数学上,模型的目标是预测富集值,以最小化不拒绝原假设的负对数似然 (NLL)。
结果
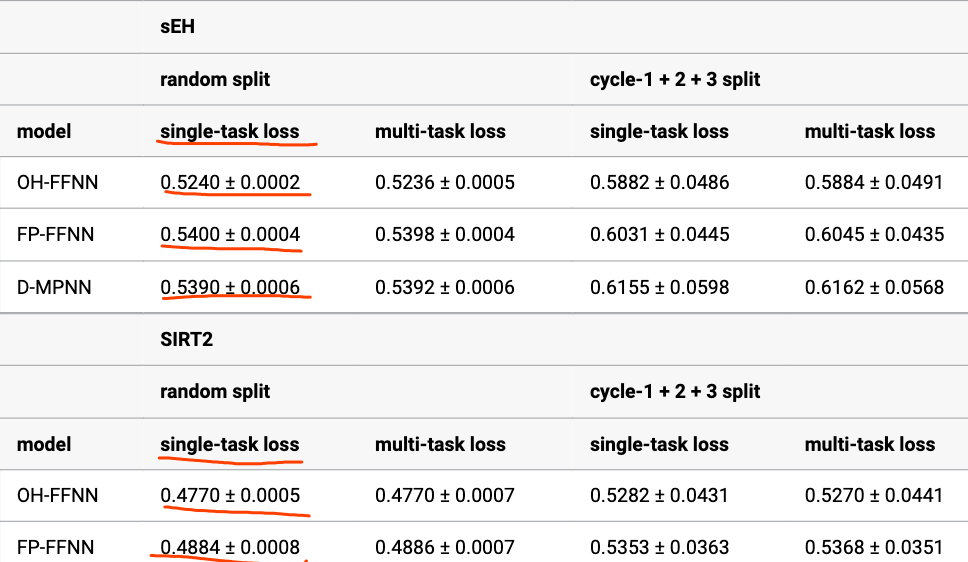
用负对数似然训练单任务模型的表现会比基于MSE训练的模型的表现有所提高
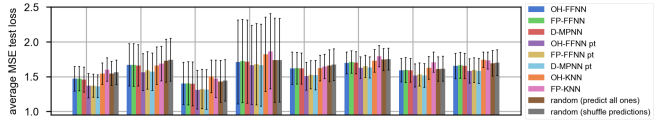
基于MSE损失的不能模型表现
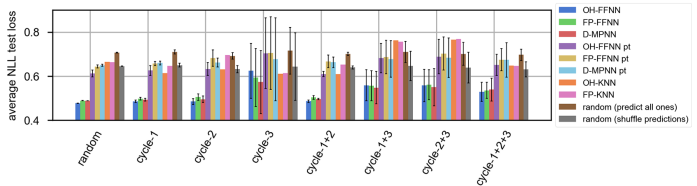
基于NLL负对数似然损失的模型表现
算法在数据CAIX上的性能显示了去噪能力并说明了计数和不确定性级别之间的联系
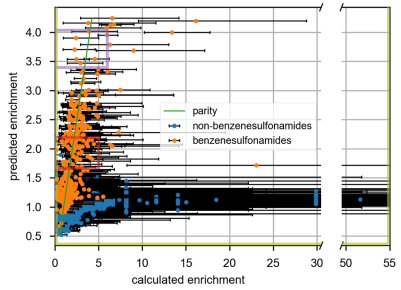
FP-FFNN在测试数据DD1S CAIX上的预测的富集值和计算的富集值之间的散点图,绿色的parity是作为参考的恒等函数,具有苯磺酰胺(benzenesulfonamide)子结构的化合物和没有苯磺酰胺(without benzenesulfonamide)子结构的化合物之间的预测富集有明显的分离,其中没有苯磺酰胺子结构的化合物是在活性位点结合CAIX锌原子的重要基序。误差线(error bars)代表计算富集的 95% 置信区间,散点图数据点的水平轴值是最大似然计算的富集值。
sHE和SIRT2数据上的性能显示了预测的富集和计算的富集之间存在粗略的相关性
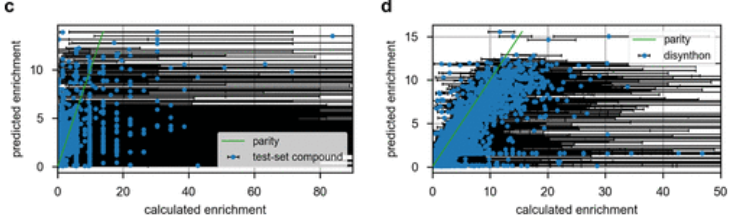
多任务模型在性能上没有显著差异
作者还研究了多任务建模是否会在提高三嗪数据集上 sEH 和 SIRT2 蛋白靶标的模型性能。在任一数据拆分上,单任务模型与多任务模型的模型性能没有显着差异,这表明负责对 sEH 和 SIRT2 的亲和力的化学特征是不同的。请注意,仍然可以考虑将这种方法扩展到更多的蛋白质靶标,以帮助预测选择性富集。
总结
作者使用泊松富集比损失函数来训练针对单个化合物的DEL数据的富集预测回归模型,这为基于DEL数据的QSAR建模引入了一种新方法。通过模型和数据之间的一致性来衡量,优化不确定性感知NLL的训练直接导致MSE损失的性能提高。
这项工作建立在 McCloskey 等人[1]的工作基础上,这表明聚合dissynthons
的二元分类可以识别具有微摩尔活性的多种类药的苗头化合物hits。作者将具有 NLL 损失函数的回归建模应用于 DEL 的单个化合物的替代方法提供了概念证明,并预计这种机器学习方法可能对 DEL 数据选择这一新兴领域的有用,以改进hits的选择,提高用于后续化合物的研究效率。
第二篇JCM文章“Machine learning on DNA-Encoded Libraries: A New Paradigm for Hit Finding"。在这项工作中,作者通过从大型商业和易于合成的化合物库中识别活性分子新方法,展示了一种将机器学习应用于 DEL 选择数据的有效性。

作者仅使用 DEL 选择数据训练模型,并将自动或可自动化过滤器应用于预测,在 30 μM 时的总命中率约为 30%,并且对于3个靶点都找到了有效的苗头化合物 (IC 50< 10 nM) 。
首先,在几种条件下对每个靶标进行亲和介导的DEL选择;然后对测序reads做处理和聚合。第三,机器学习模型在汇总的选择数据上进行了训练(不使用先前的脱DNA活性测量)并用于虚拟筛选大型文库。第四,对模型预测的最好的结果通过自动多样性过滤器、反应性子结构过滤器和化学家审查(仅限于消除具有潜在不稳定性或反应性的分子)进行过滤。最后对选定的化合物进行实验。
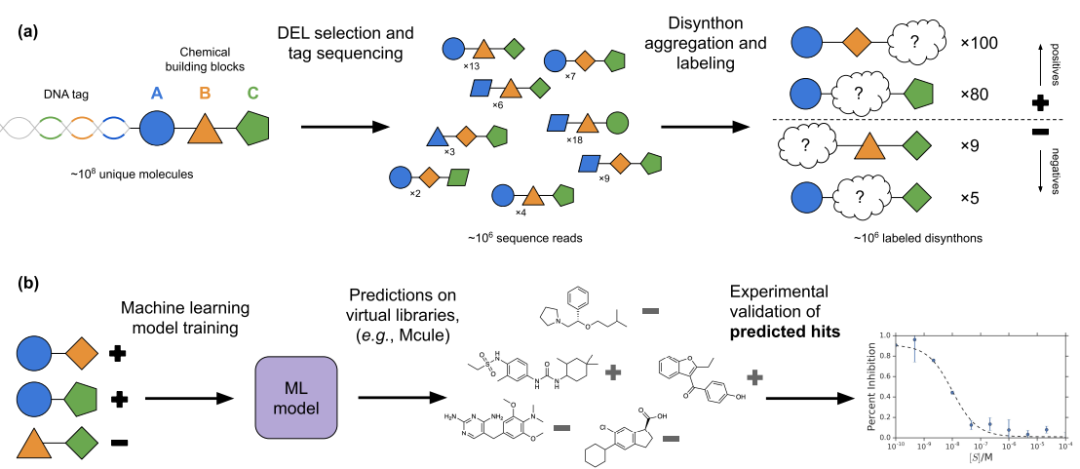
基于DEL数据训练的机器学习模型示意图。(a)从包含∼10的8次方个独立分子的DEL开始,对靶标进行亲和介导的选择,并对保留分子的DNA标签进行PCR扩增和测序。在去除PCR扩增重复后,每个库成员的reads,在共享的两循环双合子表示中聚合。基于机器学习计算的富集分数会用来对这些disynthons进行标记。对每一对可能的disynthons进行聚合(b)标记的disynthon的表征用于机器学习模型的训练数据,经过训练的模型预测来自虚拟筛选文库或商业可用catalog的先导化合物。对预测的苗头化合物进行排序和合成并进行实验测试,以确定功能测定中的活性。
作者在跨3个不同蛋白靶点(sEH,ERa,c-KIT)大概2000个化合物进行了研究。
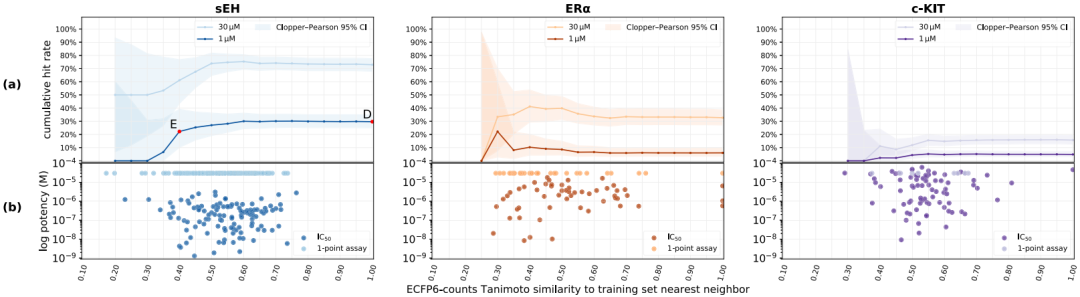
(a)GCN预测的化合物与苗头化合物的散点图(b)化合物与训练的DELs预测的化合物基于ECFP6-counts的Tanimoto相似度。累积命中率图显示了与训练集的相似性小于或等于给定(x轴)的化合物的命中率。例如,在 1 μM 时观察到的 sEH 命中率为 29.7%(sEH 的 D 点,测试了 347 种化合物),但是当仅考虑与训练集最近邻相似度≤0.40 的化合物时(E 点,测试了 36 种化合物),命中率下降到22.2%。误差带是 Clopper-Pearson 区间95% 的置信度
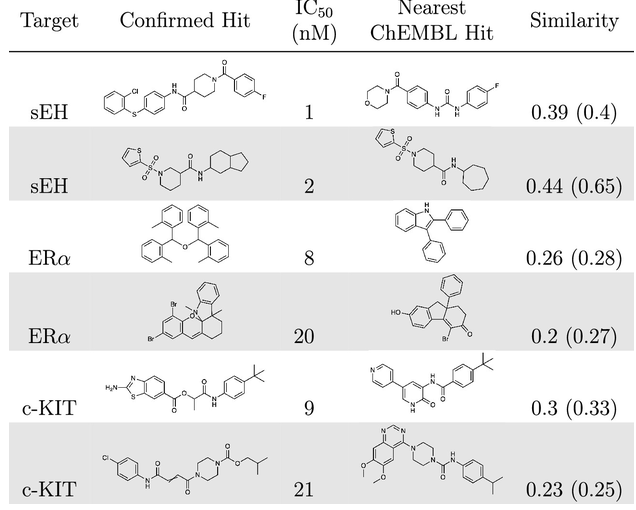
上表显示了每个靶点的一些有效苗头化合物,以及来自ChEMBL的最相似的先前已知苗头化合物。
讨论与结论
作者展示了一种新的虚拟筛选方法,该方法将 DEL 选择数据与机器学习和自动化或可自动化过滤器相结合,以发现 DEL 之外的多样化、新颖的苗头化合物。这种方法对三种不同的蛋白质靶点有效。
由于 ML 模型的泛化性,从业者在选择虚拟库方面具有很大的优势。它们可以将筛选限制在具有理想特性的分子上,例如可合成性、商业可用性、偏好子结构的存在、特定分子特性范围或与已知配体的不同的配体。在这项工作中,作者专注于可购买或易于合成的分子,这些分子往往具有类似药物的特性。这避免了将新化学物质构建到 DEL 库中并执行新选择或将新分子合并到 HTS 筛选库中的耗时且昂贵的过程。
这种考虑 DEL 之外化合物的能力是该方法的最大优势;值得注意的是,这种方法的使用成本仅为传统 DEL 筛选成本的一小部分,主要是由于合成成本的巨大差异。
这种方法的成功至少归功于三个因素:首先,过去几年出现了针对许多问题的更强大的机器学习方法。特别是对于苗头化合物的查找,作者提供了基于图的神经网络的第一个大规模前瞻性证据,它比简单的方法具有显著优势。其次,DEL 生成了大量和高质量的数据点,这对于高性能机器学习模型的训练至关重要。最后,大型商业可用文库(专有或市售)为虚拟筛选提供了低成本、结构多样的化合物来源。
参考文献
[1] McCloskey, K.; Sigel, E. A.; Kearnes, S.; Xue, L.; Tian, X.; Moccia, D.; Gikunju, D.; Bazzaz, S.; Chan, B.; Clark, M. A.; Cuozzo, J. W.; Guié, M.-A.; Guilinger, J. P.; Huguet, C.; Hupp, C. D.; Keefe, A. D.; Mulhern, C. J.; Zhang, Y.; Riley, P. Machine Learning on DNAEncoded Libraries: A New Paradigm for Hit Finding. J. Med. Chem. 2020, 63, 8857−8866.
相关代码
https://github.com/coleygroup/del_qsar
