摘要
FitzGerald, E. A. 2021. 基于片段的药物发现:识别和发展片段线索的新方法和策略。乌普萨拉科技学院学位论文数字综合摘要。59页。Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-1106-7.
在2020年,当世界面临病毒大流行时,对新药的需求变得更加明显。如何发现药物及其与社会的关系成为全世界工作场所和家庭日常讨论的一部分。因此,显然需要高效的临床前药物发现策略。
本论文的目的是通过开发基于片段的药物发现(FBDD)的新方法来促进药物发现过程,这是一种快速发展的方法,其成功依赖于获得敏感和信息丰富的分析方法以及具有合适特性的化合物。这一过程从根本上取决于科学家和工程师在生物学、化学和物理学方面的相互作用。
这个项目的特点是在一系列的研究中开发和实施新的生物物理方法,这些研究又分为。1. 为具有挑战性的目标开发生物传感器检测和方法,2.利用生物传感器发现针对动态蛋白质的片段,以及3. 利用基于片段的策略重建配体。
选择不同的目标作为本文所述的目标不可知方法的挑战原型。重点目标是:乙酰胆碱结合蛋白(AChBP),一种配体门控离子通道的可溶性同源物,以及两种复杂的多域表观遗传酶赖氨酸特异性脱甲基酶1(LSD1)和SET和含MYND域的蛋白3(SMYD3)。在建立强大的筛选策略之前,需要对蛋白变体进行表达、纯化、工程化和生化表征。
基于不同的时间分辨和非常敏感的检测原理(SPR、SHG、GCI)的三种类型的生物传感器被用来识别和描述蛋白质的新片段的相互作用动力学。在SPR方面,为筛选针对困难目标的片段设计了各种复用检测方法。值得注意的是,它导致了SMYD3中异生配体和位点的确定,随后利用X射线晶体学对其进行了动力学和结构上的表征,并利用计算方法进一步发展。
开发了一种创新的SHG检测方法,用于特异性检测诱导构象变化的配体,并用于针对AChBP的片段筛选。结果显示,使用这种技术可以鉴定出有可能作为配体门控离子通道功能调节器的片段。新的生物物理和计算方法的联合应用使我们能够为药物发现项目确定有用的起点。
关键词:生物化学、药物发现、生物物理学、基于片段的药物发现、表观遗传学、生物传感器、表面等离子体共振、相互作用分析、第二谐波
1 简介
1.1 药物发现
在撰写本报告时,世界正面临着有史以来最具破坏性的大流行病之一。新型冠状病毒SARS-CoV-2对人类生活产生了毁灭性的影响,造成数百万人死亡,给全球经济带来严重后果。讨论药物发现的重要性和基本原理从未像现在这样恰当和及时。这些项目是如何启动的,需要什么,以及2021年的科学如何帮助我们加速发现?尽管药物的定义在一千年里可能已经发生了变化,但这个概念却远非新鲜事物。几千年来,服用灵丹妙药、药水或药物一直以这种或那种形式存在着。这方面的证据散落在历史书中。无论是公元前2700年中国从简陋的茶叶中提取咖啡因,还是动物食用天然产品的镇痛作用,例如大猩猩咀嚼古柯叶以抑制饥饿和缓解疼痛。
快进到1855年,我们看到通过科学技术的进步和科学干预,德国化学家Fredrich Gaedcke从古柯叶中分离出可卡因生物碱,将其命名为 "红霉素"。正是在19世纪末和20世纪初,我们看到了现代制药业的发展,它主要是在瑞士和北美的石化染料工业和精细化学工业的基础上进行的。然而,真正的进步来自于基于结构或以结构为导向的药物发现,也就是现在所称的早期药物发现(ESDD)或临床前药物发现。
1.2 早期阶段和临床前药物发现
基于结构的药物发现(SBDD),通常被称为合理药物设计,是一种利用已知药物的化学结构或感兴趣的目标结构的预先知识来合理设计或指导过程的方法。这方面的一个例子是,在发现抗生素青霉素后,科学家们通过生产半合成的β-内酰胺化合物,并在β-内酰胺环周围进行各种化学修饰,迅速生产出母分子的后续变体。这导致发现了更有效的和/或不易产生抗生素抗性的药物。随着技术的进步和科学方法的改进,人们把重点放在了解感兴趣的目标的三维结构的结合袋上。由于重组蛋白生产和结构阐明方法(即X射线晶体学(XRC)和核磁共振(NMR))的进步,实验人员能够直观地了解化合物与目标蛋白的结合模式。这为使用计算方法提供了可能性,即通过补充结合袋的形状和电荷来可视化、建模和改进未来的化合物迭代。药物发现管道中的这一过程(图1)通常被统称为 "临床前发现",其中包括目标验证、命中率识别和线索生成。一旦一个目标被优化,它就会进入临床阶段。
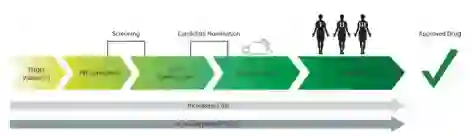
图1. 药物发现管道的示意图。随着流程从一个阶段进入下一个阶段,成本和失败的风险都会增加。
1.2.1 临床前药物发现的各个阶段
目标验证、筛选、命中率生成和线索优化是相互关联的。在这些步骤中,经常会发生从一个步骤到另一个步骤的迭代运动。进展到下一步的一个关键点是使用基于不同检测原理的方法对化合物进行正交验证。这个过程可以被看作是一个决策工作流程,总结在图2中。
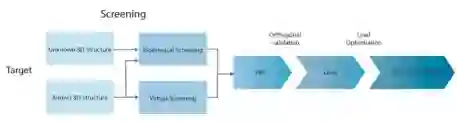
图2. 从验证的靶点到临床前线索或临床候选药物的决策工作流程。如果蛋白质的三维结构是已知的,那么在用生物物理方法验证之前,可以在硅学中对化合物库进行虚拟筛选。
1.2.2 高通量筛选
在九十年代初,大的制药公司大量投资于高通量筛选(HTS)的巨大基础设施。这包括建立几十万甚至上百万的化合物库,并建立先进的机器人技术来处理所需的吞吐量。一个典型的HTS库的特点是药物或类似铅的化合物,它们遵循Lipinski的59规则,分子量小于500 Da。理论上,这些化合物应该具有显示高亲和力相互作用的潜力,即对各种感兴趣的目标具有低微摩尔到纳摩尔范围的KD值。在大多数情况下,建立一个非常敏感的生化检测方法来快速筛选这些非常大的库。如果化合物符合检测的标准,例如对目标的抑制,它们就被认为是命中的。在对命中的化合物进行验证后,从而排除了由于实验伪影(假阳性)而出现的命中的化合物,对这些化合物进行结构优化,以改善其理化和生物学特性。图3说明了一个典型的HTS方法。

图3. 代表一个典型的HTS方法的插图。一个感兴趣的目标以绿色显示,HTS库以相互连接的形状定义。尽管化合物符合假设的结合口袋,但没有什么优化的空间。
无论这些检测方法多么强大,它们往往由于许多原因而显得不足。1. 尽管文库可以包含数以百万计的化合物,但它们往往缺乏新目标所需的复杂性,而且只探测了可用化学空间的一小部分。2. 2.这些化合物通常是以前项目的残留物,可能缺乏新目标类别所需的特异性。3. 3. 由于化学成分和类似于铅或药物,它们可能限制了进一步优化的灵活性。尽管这些化合物很容易被识别出来,但它们往往以大的疏水分子为特征,导致 "油腻 "的线索,具有不利的药代动力学特征。葛兰素史克公司的Mike Hann认为,这些化合物患有 "分子肥胖症 "10,即大体积的化合物,由于其疏水性,在未来的候选提名中存在风险,几乎没有优化的余地。因此,这种类型的优化所产生的分子往往在ADME谱上有问题,如溶解度和口服生物利用度。
1.2.3 基于片段的药物发现
其核心原则和理论是建立在生物物理检测的高灵敏度和高通量的基础上,从核磁共振开始,随后是X射线晶体学和SPR,这些方法不断发展,并具有覆盖足够大的化学空间的片段库所需的通量。在2000年初,FBDD从一个小众技术变成了一门合法的学科,迅速被工业界和学术界采用。
这种牵引力是由于实验设计的进步和生物物理仪器灵敏度的提高。不是依靠大量的化学文库,而是把重点放在把文库的复杂性减少到化合物的片段上。从理论上讲,这种简化的HTS方法表明,通过使用结构多样、分子量较低的化合物,可以更有效地探测更广泛的化学空间,而不是传统的HTS或类药物化合物库中的化合物。
对于FBDD,库通常遵循 "三原则 "的概念,即片段的分子量<300,cLogP≤3,氢键供体的数量≤3,氢键受体的数量≤3。由于规模较小,人们可以通过探索片段连接/合并或增长的策略来探索更大的化学空间(图4)。这也为以后优化线索生成提供了灵活性,包括在候选提名阶段,当不同的理化性质被调整以预测候选药物的有利的ADME特性时。
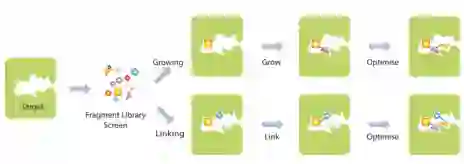
图4. 典型的针对感兴趣的靶点(绿色)的FBDD筛选,以及片段库(彩色的形状)的说明。在结构和药物化学路线的指导下,一个片段可以结合并1. 如果发现两个片段彼此相近,它们可以被连接起来。在这两种情况下,化合物都被优化以利于ADME和生物利用度。
如前所述,药物发现过程往往是一个漫长的、有风险的和昂贵的业务。尽管这一事实,我们已经看到许多新型疗法进入后期临床试验的例子,更重要的是,到目前为止,有四种FDA批准的药物是通过基于片段的方法发现的:vemurafenib17, erdafitinib18, venetoclax19和pexidartinib20(图5)。
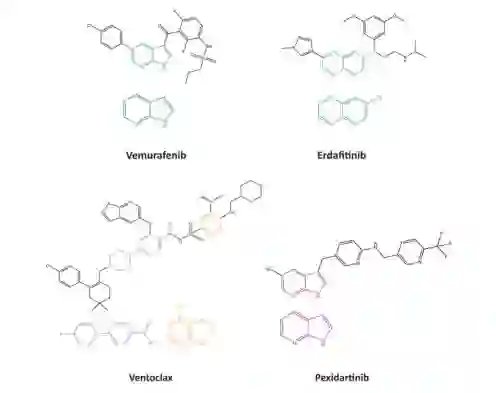
图5. 目前以片段为基础的方法开发的药物,以及由此产生的初始片段命中。
1.2.4 跨学科和高度合作的努力
FBDD依赖于许多学科之间的紧密互动。一个项目要想成功,没有一个科学家或一个学者能够现实地期望领导一个从目标识别到上市的药物的活动。在临床前阶段,从成功到领先所需的检测和专业知识是非常广泛的。有了这些知识,为了解决可能被认为是一项巨大的任务,工业界和学术界已经看到了合作的力量。
由于FBDD需要高度专业的技能,因此在玛丽-斯科沃多夫斯卡-居里行动(MSCA)下建立了一个泛欧洲项目,以培训FBDD方面的早期职业研究人员。该项目被称为FRAGNET,即片段网络,通过培训与FBDD相关的所有方面的研究人员,促进跨学科研究。研究人员被安排在工业或学术主办机构,专门从事FRAGNET联盟定义的四个支柱之一(图6)。1. 库 2. 筛选 3. 设计,和4. 优化。例如,参与开发生物物理检测的研究人员将依赖于联盟开发的文库,同样,参与设计的研究人员将依赖于合作,并接受开发筛选检测和后续线索优化的合作伙伴的指导。这种相互作用将在本论文的整个过程中变得明显。
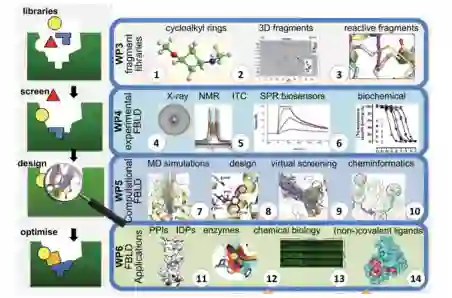
图6. FRAGNET战略概述,包括在网络中进行的FBDD的四个支柱:1.库 2.筛选 3.设计 4.优化。