
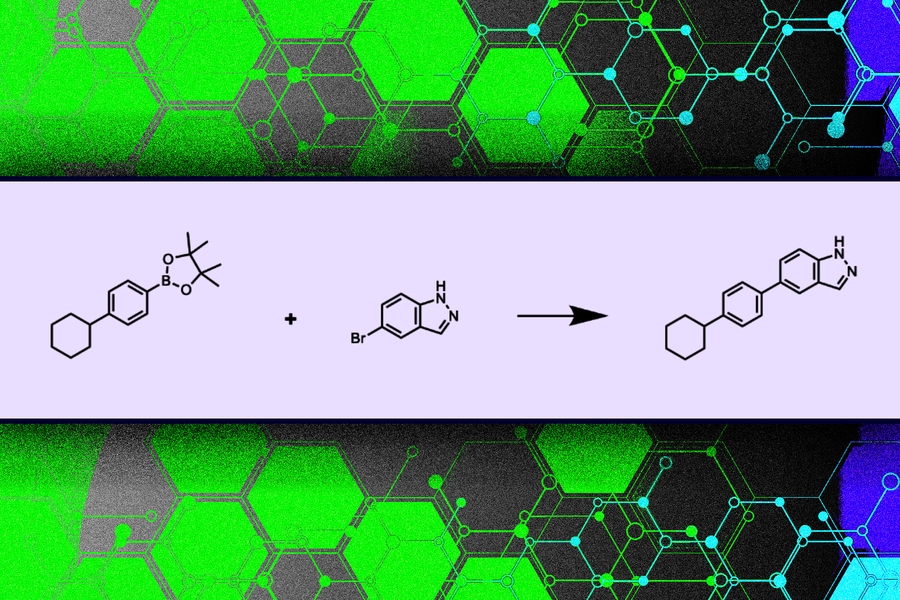
麻省理工学院的研究人员开发了一种机器学习模型,该模型为药物发现过程提出了新的分子,同时确保它所建议的分子实际上可以在实验室中合成。
背景
制药公司正在使用人工智能来简化发现新药的过程。机器学习模型可以提出具有特定性能的新分子,这些分子可以对抗某些疾病,在几分钟内完成人类可能需要几个月才能手动完成的工作。
但是有一个主要障碍阻碍了这些系统的发展:这些模型通常会提出在实验室中难以或不可能生产的新分子结构。如果化学家实际上不能制造这种分子,则无法测试其抗病特性。
麻省理工学院研究人员的一种新方法改进了机器学习模型,该模型只建议可以合成的分子结构。该方法保证分子由可以购买的材料组成,并且这些材料之间发生的化学反应遵循化学定律。
与其他方法相比,此模型提出的分子结构在流行的评估中得分一样高,有时甚至更好,并保证可合成。他们的系统需要不到一秒钟的时间来提出合成途径,而其他模型提出分子结构然后评估其可合成性的方法可能需要几分钟。
“这个过程重新定义了如何要求这些模型生成新的分子结构。这些模型中的许多都考虑逐个原子或逐个键地构建新的分子结构。取而代之的是,我们正在逐个构建新的分子构件,并通过反应构建新的分子,”该论文的作者,麻省理工学院化学工程、电气工程和计算机科学系的助理教授 Connor Coley 说。
与Coley一起撰写该论文的还有第一作者研究生Wenhao Gao和博士后Rocío Mercado。这项研究将在本周的国际学习表征会议(ICLR)上发表。
构建模块
为了创建分子结构,该模型模拟了合成分子的过程,以确保它可以被生产出来。
该模型给出了一组可行的构建块,它们是可以购买的化学物质,以及可以使用的有效化学反应列表。这些化学反应范本由专家手工制作。通过只允许某些化学物质或特定反应来控制这些输入,研究人员可以限制新分子的搜索空间大小。
该模型使用这些输入来构建一棵树,方法是选择构建块并通过化学反应将它们连接起来,一次一个,构建最终的分子。在每个步骤中,随着添加额外的化学物质和反应,分子会变得更加复杂。
它输出最终的分子结构以及合成它的化学和反应树。
“我们没有直接设计产品分子本身,而是设计了一个动作序列来获得该分子。这使我们能够保证结构的质量,”Gao说。
为了训练他们的模型,研究人员输入了一个完整的分子结构和一组构建模块和化学反应,模型学习创建一个合成分子的树。在查看了数十万个示例后,该模型学会了自己提出这些合成路径。
分子优化
训练后的模型可用于优化。研究人员定义了他们希望在最终分子中实现的某些特性,给定某些构建块和化学反应范本,并且该模型提出了可合成的分子结构。
“令人惊讶的是,你可以用如此小的范本集实际复制大部分分子。不需要那么多构建块来生成大量可用的化学空间供模型搜索,”Mercado 说。
他们通过评估模型重建可合成分子的能力来测试该模型。它能够复制 51% 的这些分子,并且只需不到一秒钟的时间就可以复制每个分子。
他们的技术比其他一些方法更快,因为模型没有搜索树中每个步骤的所有选项。Gao解释说,它有一套明确的化学物质和反应可供使用。
当使用模型提出具有特定性质的分子时,他们的方法提出了更高质量的分子结构,这些分子结构比其他方法具有更强的结合亲和力。这意味着这些分子将能够更好地附着在蛋白质上并阻止某种活动,例如阻止病毒复制。
例如,当提出一种可以与 SARS-Cov-2 对接的分子时,他们的模型提出了几种分子结构,这些分子结构可能比现有的抑制剂更能与病毒蛋白结合。然而,正如作者承认的那样,这些只是计算预测。
“有很多疾病需要解决,”Gao说。“我希望我们的方法可以加速这一过程,这样我们就不必每次都筛选数十亿个分子来寻找疾病目标。相反,我们可以只指定我们想要的特性,它可以加速寻找候选药物的过程。”
他们的模型还可以改善现有的药物发现管道。Mercado 说,如果一家公司已经确定了一种具有所需特性但无法生产的特定分子,他们可以使用该模型来提出与其非常相似的可合成分子。
现在他们已经验证了他们的方法,该团队计划继续改进化学反应范本,以进一步提高模型的性能。借助额外的范本,他们可以对某些疾病目标进行更多测试,并最终将模型应用于药物发现过程。
“理想情况下,我们需要能够自动设计分子并同时快速为我们提供合成树的算法,”参与了这项工作的微软剑桥研究院(英国)的 Marwin Segler 说。。“Coley 教授和团队的这种优雅方法是解决这个问题的重要一步。虽然早期有通过合成树生成进行分子设计的概念验证工作,但这个团队确实做到了。他们第一次在有意义的规模上展示了出色的性能,因此它可以对计算机辅助分子发现产生实际影响。这项工作也非常令人兴奋,因为它最终可以为计算机辅助合成规划提供新的范式。这可能会对该领域的未来研究产生巨大的启发。”
这项研究得到了美国海军研究办公室和药物发现与合成机器学习联盟的部分支持。
论文

分子设计和合成规划是分子发现过程中的两个关键步骤,本文建议将其表述为有条件合成途径生成的单一共享任务。提出一种摊销的方法,将合成途径生成为以目标分子嵌入为条件的马尔科夫决策过程。该方法使得能以自下而上的方式进行合成规划,并通过对优化条件码进行解码来设计可合成的分子,证明了同时解决设计和合成问题的潜力。该方法利用神经网络对合成树进行概率建模,一次一个反应步骤,根据反应模板的离散行动空间中编码的反应性规则。对从可购买的化合物库和专家策划的模板列表中产生的数十万条人工途径进行了训练。用以下方法来验证该方法:(a)利用条件生成恢复分子;(b)识别可合成的结构类似物;(c)优化与药物发现有关的分子结构。




