
最新AI+药物设计论文

基于结构的药物设计(Structure-based drug design,简称SBDD),利用蛋白质的三维几何结构来识别潜在的药物候选物,正逐渐成为药物发现过程中的重要环节。然而,传统的基于物理化学建模和专家领域知识的方法既耗时又劳动密集。近期,几何深度学习的进步(它整合和处理3D几何数据)以及像AlphaFold这样的工具提供的准确蛋白质3D结构预测的可用性,已经显著推动了基于结构的药物设计的发展。在本文中,我们系统地回顾了几何深度学习在基于结构的药物设计方面的最新进展。我们首先简要讨论了基于结构的药物设计中的主流任务,常用的3D蛋白质表示法,以及具有代表性的预测/生成模型。然后,我们深入探讨了每个任务(包括结合位点预测,结合姿态生成,去新分子生成,连接片设计和结合亲和力预测)的详细评论,包括问题设置,具有代表性的方法,数据集和评估指标。最后,我们总结本文,并讨论了几何深度学习在基于结构的药物设计方面面临的当前挑战,并强调了其潜在机会。
https://www.zhuanzhi.ai/paper/36919c87375cfe74d4cf8e4330da3094
1. 引言
基于结构的药物设计(Structure-based drug design,简称SBDD)[1],[2],[3]通过有效利用目标蛋白质的三维几何信息来设计和优化药物候选物,正成为一种必不可少的工具。传统上,目标蛋白质的3D结构是通过像X射线晶体学[4]、核磁共振(NMR)光谱学[5]或冷冻电子显微镜(cryoEM)[6]这样的技术获得的。最近,像AlphaFold[7]这样的高精度蛋白质结构预测的进步进一步增加了结构数据的可用性,并为其广泛应用奠定了基础。制药公司和科学家广泛使用SBDD。已成功通过SBDD识别了一些上市药物,如HIV1蛋白酶抑制剂[8],胸腺苷酸合成酶抑制剂raltitrexed[9]和抗生素norfloxacin[10]。然而,依赖物理建模、手工打分函数和枚举的传统基于结构的药物设计方法仍然耗时且劳动密集。为解决这些问题,最近提出了几何深度学习[11],以加速和改进基于结构的药物设计过程。
几何深度学习(Geometric Deep Learning,简称GDL)[11]是一种融合和编码3D几何数据的神经网络架构。它们可以自动提取信息丰富的3D结构特征,而无需手动特征工程。此外,一些GDL方法,如EGNN[12],将对称性属性纳入网络设计,作为一种有效的归纳偏差以获得更好的性能。在3D欧几里得空间的背景下,对称性涉及旋转,平移和反射,以及蛋白质和分子性质在这些变换下是如何改变的(在第2.3节有更多讨论)。随着几何深度学习的迅速发展,一系列SBDD任务,包括结合位点预测[13],结合姿态生成[14],去新分子生成[15],连接片设计[16]和结合亲和力预测[17]已经受益。总的来说,几何深度学习在SBDD领域的发展速度非常快,吸引了越来越多的关注。
首先,在第2.2节中,我们简要介绍了蛋白质的主流3D表示、本文审查的SBDD任务,以及流行的预测和生成模型。接下来的章节按照SBDD任务的逻辑顺序组织,如图1所示:对于目标蛋白质结构,我们首先需要确定结合位点;然后我们可以进行结合姿态生成、去新分子生成和连接片设计;有了蛋白质-配体复合物结构,我们可以使用结合亲和力预测和其他筛选标准来筛选药物候选物。诚然,SBDD任务的顺序和类别并不固定,因为SBDD是一个迭代过程,通过多个周期使优化的药物候选物进入临床试验[24]。一些方法也可能能够完成多个任务。例如,EquiBind[25]可以在不事先了解结合位点的情况下预测配体的结合姿态,即盲底物对接。为了便于读者理解。最后,第4节确定了开放的挑战和机遇,为未来设计用于基于结构的药物设计的几何深度学习方法铺平了道路。
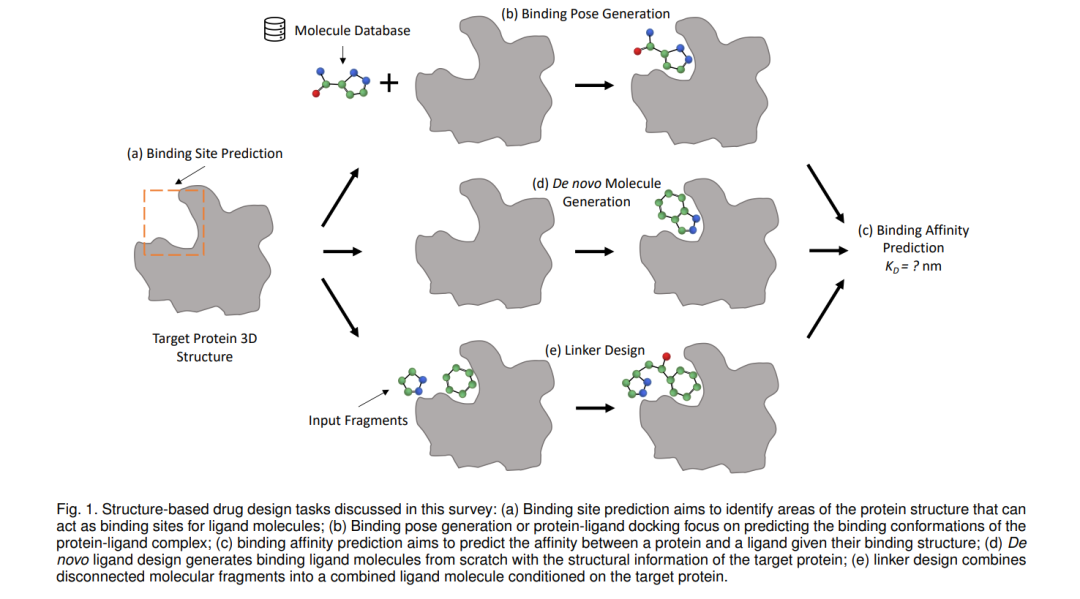
2 结构基药物设计任务概述
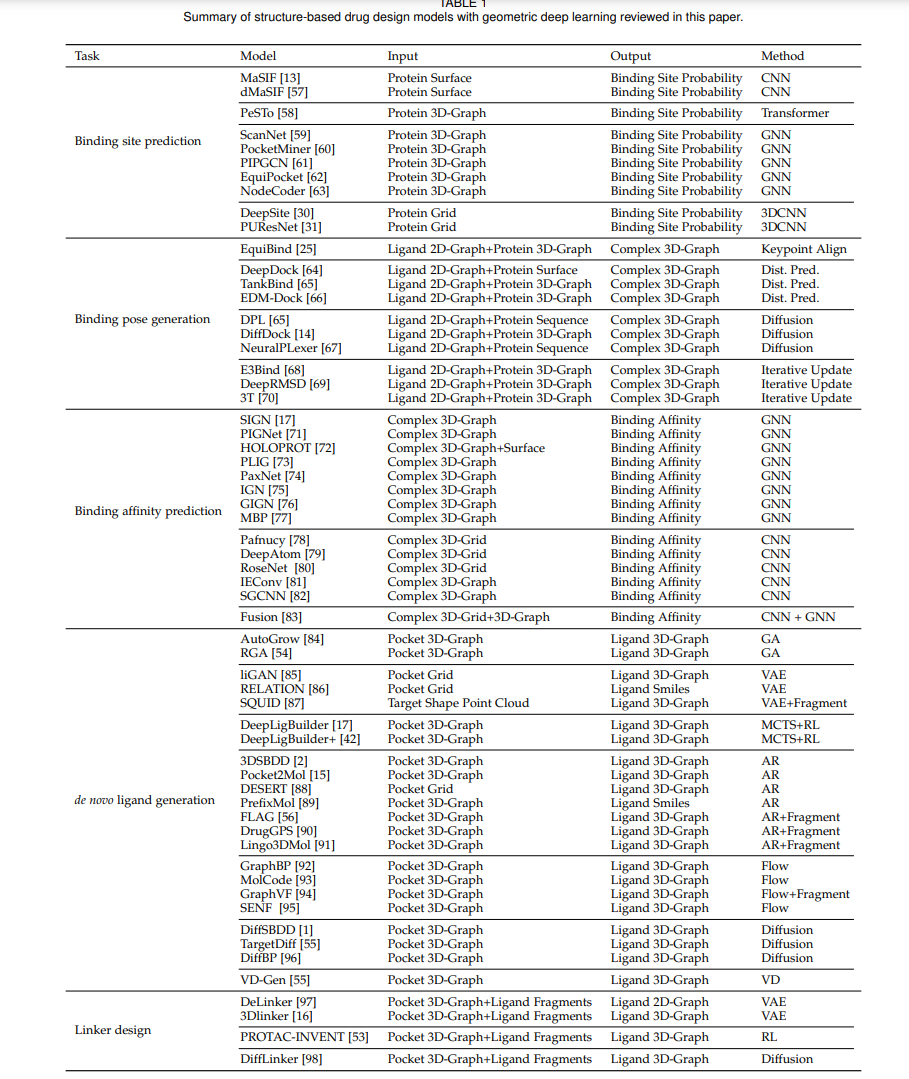
在图1中,我们展示了本论文中审查的结构基药物设计任务的概述,包括结合位点预测、结合位姿生成、全新分子生成、连接物设计和结合亲和力预测。一般来说,结合位点预测和结合亲和力预测是预测任务。其他任务是利用生成模型来处理的生成任务。在这里,我们总结了预测任务(即结合位点预测和结合亲和力预测)的主流预测方法。这些模型也广泛用作生成方法的结构编码骨干。我们总结了结构基药物设计任务中流行的生成方法,包括自回归模型、流模型、变分自编码器、扩散模型以及其他一些方法。
- STRUCTURE-BASED DRUG DESIGN的三个任务
3.1 结合位点预测
蛋白质表面指的是与环境相互作用的蛋白质外部区域,通常以具有几何和化学特征的连续形状表示。基于蛋白质表面表示的结合位点预测为其他结构基药物设计任务(如结合位姿生成和全新配体生成)奠定了基础。形式上,将目标蛋白质表面表示为S(例如,网格/点云),目标是学习一个预测模型f(S),该模型输出表面上每个点是结合位点的概率。分子表面相互作用指纹(MaSIF)[13]方法首创了使用基于3D网格的几何深度学习来预测蛋白质相互作用位点。然而,MaSIF受到预计算网格、手工设计特征和大量计算时间的限制。dMaSIF [57]对MaSIF进行了扩展,并提出了一种基于蛋白质的3D点云表示的高效端到端预测框架。此外,一些最近的工作将蛋白质表面建模为3D图,并设计了基于GNN [59]或图转换器的方法 [58],以实现高效且准确的结合位点预测。蛋白质数据银行(Protein Data Bank,PDB)[99]包含通过X射线晶体学、NMR光谱学或冷冻电子显微镜等方法获得的3D结构蛋白质数据。Dockground [100]提供了从PDB中提取的一组全面的蛋白质相互作用复合物数据。由于通常不存在分类的阈值,ROCAUC常用于评估结合位点预测模型的性能。
3.2 结合位姿生成
预测配体分子如何与目标蛋白质结合是药物发现中的一个基本问题,广泛应用于虚拟筛选和药物工程领域。形式上,将目标蛋白质结构表示为P,配体分子的2D图表示为G,配体的3D结构表示为R,目标是学习一个模型p(R|G,P),用于预测配体的3D结构。传统的对接方法依赖于手工设计的评分函数和不充分的构象采样。最近,几何深度学习方法被用于解决对接问题,并取得了令人瞩目的进展。例如,EquiBind [25] 是一个SE(3)-等变几何深度学习模型,通过预测和对齐配体和蛋白质上的口袋关键点,直接预测受体结合位置(盲对接)和配体的结合位姿。TANKBind [65] 在EquiBind的基础上进行了改进,通过独立预测每个可能的口袋的对接位姿(原子间距离矩阵),然后对其进行排序。此外,TANKBind提出了一种考虑三角函数约束的神经网络用于结合结构预测。与预测距离矩阵不同,DiffDock [14] 在配体位姿空间上设计了一个扩散生成模型,用于分子对接;E3Bind [68] 则使用等变网络迭代地更新配体位姿。然而,蛋白质的结构是灵活的,可以发生内在或诱导的构象变化[101],这些方法忽略了这一点。一些最近的方法,如3T [70],进一步考虑了结合蛋白质的灵活性,在蛋白质-配体复合物结构预测上取得了更优越的性能。
3.3 结合亲和力预测
蛋白质-配体结合亲和力是一种衡量相互作用强度的指标,是一项回归任务。准确的亲和力预测有助于设计有效的药物分子,在结构基础药物设计中发挥着重要作用。形式上,将已结合的蛋白质结构表示为P,已结合的配体表示为L,结合亲和力表示为y,我们的目标是训练一个模型 f(P,L) = y 用于结合亲和力的预测。结合亲和力预测方法的探索具有悠久的历史。早期的研究主要利用经验公式[103]或设计手工特征结合传统机器学习算法进行结合亲和力预测[104]。尽管取得了一些进展,但这些方法在预测准确性方面仍然存在局限性,并需要在特征设计方面投入大量的努力。最近的研究强调了几何深度学习方法的应用,这些方法将蛋白质-配体复合物的结构表示为3D网格或3D图进行处理和预测。这些方法直接使用CNN或GNN对复合物的3D结构与结合亲和力之间的关系进行建模。例如,给定一个复合物结构,Pafnucy [78] 提取以配体几何中心为焦点的20 A˚立方盒子,并将其离散化为一个分辨率为21 × 21 × 21 × 19、边长为1 A˚的网格。然后使用3D-CNN处理该网格,将其视为多通道的3D图像。SIGN [17]将复合物结构转换为复杂的3D图,并设计了一种结构感知的交互图神经网络,使用基于极坐标的图注意力层以半监督的方式捕捉3D空间信息和全局长程相互作用。Fusion [83]同时利用复杂的3D网格表示和3D图来捕捉不同相互作用的特征。HOLOPROT [72]考虑了复合物的结构和表面。MBP [77]引入了第一个亲和力预训练框架,其中涉及训练模型来预测来自相同生物测定的样本排序。这种预训练是使用自建的ChEMBL-Dock数据集进行的,该数据集包含超过300,000个实验亲和力标签和约2.8M个通过对接生成的复合物结构。总体而言,基于几何深度学习的方法能够有效捕捉3D结构信息,并展现出卓越的预测准确性。
3.4 从头设计的配体生成
de novo ligand generation 的目标是生成适应并与特定的蛋白质结合位点发生作用的有效的三维分子结构。de novo 表示在没有给定参考配体分子的情况下生成分子,即从头开始生成分子。形式上,假设 P 表示蛋白质结构,G 表示三维配体分子,目标是学习一个条件生成模型 p(G|P) 来捕捉蛋白质-配体对的条件分布。 在 de novo 配体生成的早期工作中,将目标蛋白质表示为三维网格,并采用三维卷积神经网络作为编码器。例如,LiGAN [109] 使用基于原子密度网格表示的蛋白质-配体结构的条件变分自编码器进行配体生成。然后,通过从生成的原子密度进一步进行原子拟合和键推断来构建配体的分子结构。作为初步工作,LiGAN 并不满足理想的等变性质。后续的工作将目标蛋白质和配体表示为三维图形/点云,并通过利用图神经网络进行上下文编码来实现等变性。例如,3DSBDD [110] 使用 SchNet [111] 来编码结合位点的三维上下文,并估计原子在三维空间中出现的概率密度。原子会被自回归地采样,直到没有空间容纳新的原子为止。GraphBP [112] 采用了归一化流 [113] 的框架,并构建局部坐标系来预测原子类型和相对位置。Pocket2Mol [15] 采用了几何向量感知器 [114] 和基于向量的神经网络 [115] 作为上下文编码器。它还明确考虑和预测共价化学键。通过利用分子片段的化学先验知识,如功能团,FLAG [56] 和 DrugGPS [90] 提出了逐片段生成配体分子的方法,并产生更真实的亚结构。最近,受到扩散模型强大的生成能力的启发,提出了基于扩散的方法,如 DiffSBDD [1] 和 TargetDiff [55],用于非自回归的配体生成,并取得了更优越的性能。
3.5 连接物设计
大多数小分子药物通过与靶蛋白结合并抑制其活性来发挥作用。然而,由于疾病的复杂性,目标蛋白中的一些氨基酸可能发生突变,导致结合亲和力减弱,药物脱落。为了解决这个问题,最近开发了一种名为蛋白酶靶向嵌合体(PROTAC)的药物机制,通过促使目标蛋白的完全降解来抑制蛋白功能。具体而言,PROTAC 包含两个分子片段和一个将这些片段连接成完整分子的连接物。PROTAC 中的一个片段与目标蛋白结合,另一个片段与能够降解目标蛋白的另一种分子结合。由于PROTAC 只需要对其靶点具有高选择性,而无需抑制目标蛋白的活性,因此重点放在将先前无效的抑制剂分子重新用作下一代药物的REPURPOSING 上。PROTAC 中的一个关键问题是连接物的设计,它在给定分子片段和目标蛋白的条件下生成连接物。形式上,将目标蛋白表示为P,分子片段表示为F,连接物表示为L,目标是学习一个条件生成模型p(L|F,P)。 DeLinker [97] 使用变分自编码器(VAE)架构,并自回归地生成连接物的原子和边。DeLinker 只考虑了简单的几何信息,如相对距离和方向,并输出二维分子图。3Dlinker [16] 进一步可以生成三维分子结构,并且不需要指定锚定原子,即片段的连接原子。PROTAC-INVENT [53] 同时训练强化学习算法,以使生成的PROTAC结构倾向于具有预定义的二维和基于三维的属性。然而,连接物的设计取决于目标蛋白的结构信息,在上述方法中被忽略了。DiffLinker [98] 设计了一个条件扩散模型,根据目标蛋白的结构为一组输入的分子片段生成分子连接物。结果表明,考虑目标蛋白的结构可以提高生成的PROTAC分子的结合亲和力。
4 结论
在本综述中,我们系统地回顾了基于几何深度学习的结构药物设计。我们根据任务将现有的工作分为五个类别进行组织。对于每个任务,我们提供了问题的表述,总结了一系列代表性的工作,并列出了用于评估的数据集和指标。最后,我们确定了结构药物设计领域的挑战和机遇。尽管每年都有大量的新方法和技术被提出,但我们希望本综述能帮助读者快速了解当前方法的局限性,并为几何深度学习模型在结构药物设计领域的未来铺平道路。
