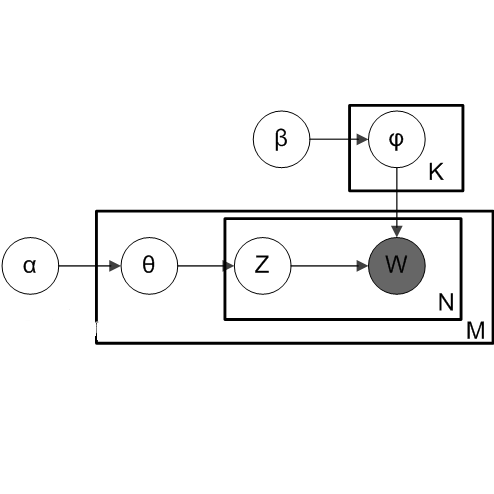
Mixed-membership (MM) models such as Latent Dirichlet Allocation (LDA) have been applied to microbiome compositional data to identify latent subcommunities of microbial species. However, microbiome compositional data, especially those collected from the gut, typically display substantial cross-sample heterogeneities in the subcommunity composition which current MM methods do not account for. To address this limitation, we incorporate the logistic-tree normal (LTN) model -- using the phylogenetic tree structure -- into the LDA model to form a new MM model. This model allows variation in the composition of each subcommunity around some ``centroid'' composition. Incorporation of auxiliary P\'olya-Gamma variables enables a computationally efficient collapsed blocked Gibbs sampler to carry out Bayesian inference under this model. We compare the new model and LDA and show that in the presence of large cross-sample heterogeneity, under the LDA model the resulting inference can be extremely sensitive to the specification of the total number of subcommunities as it does not account for cross-sample heterogeneity. As such, the popular strategy in other applications of MM models of overspecifying the number of subcommunities -- and hoping that some meaningful subcommunities will emerge among artificial ones -- can lead to highly misleading conclusions in the microbiome context. In contrast, by accounting for such heterogeneity, our MM model restores the robustness of the inference in the specification of the number of subcommunities and again allows meaningful subcommunities to be identified under this strategy.
翻译:混合成员(MM) 模型,如Lentant Dirichlet 分配(LDA) 模型,已经应用到微生物组成数据中,以识别微生物物种的潜在亚群。然而,微生物组成数据,特别是从肠内收集的数据,通常显示目前MM方法不考虑的亚群构成中存在大量交叉抽样差异。为解决这一限制,我们将物流-树正常(LTN)模型(使用植物遗传树结构)纳入LDA模型,以形成一个新的MDA模型。这一模型允许每个子群的组成围绕某种“中心机器人”的构成的对比度变化。纳入辅助 P\'olya-Gamma变量,使得一个计算高效的封闭Gibs取样器能够在这个模型下传递Bayes的变异性。我们比较新的模型和LDA,并表明,在大型交叉抽样(LDAA模型)存在的情况下,导致的推论可以非常敏感到子群落的子群落的规格,因为这个模型并没有将这种结果转化为战略中的精确性。