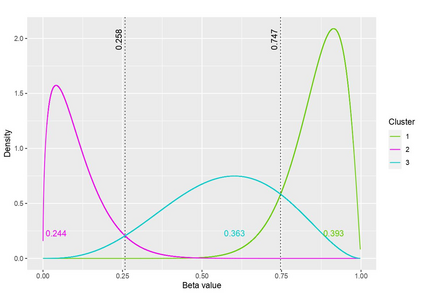
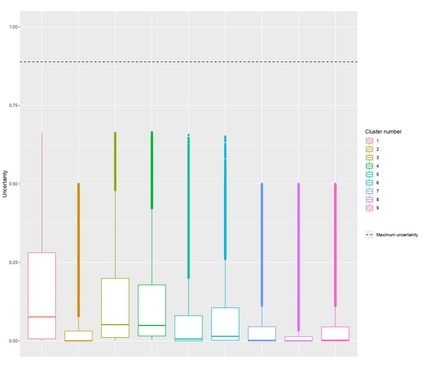
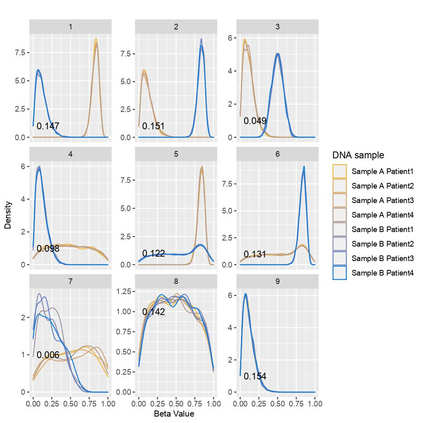
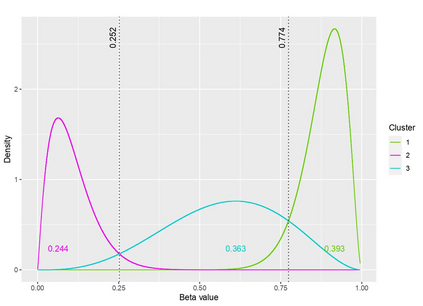
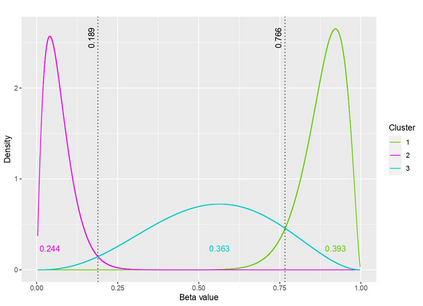
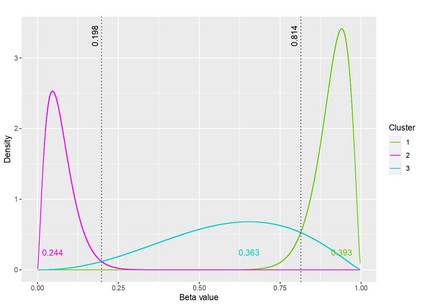
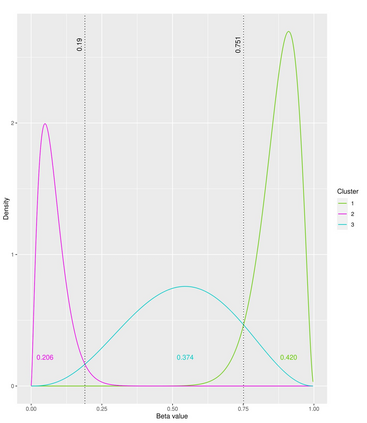
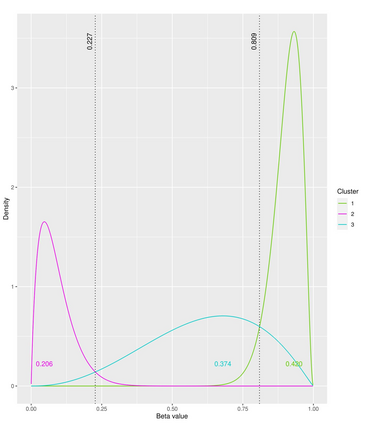
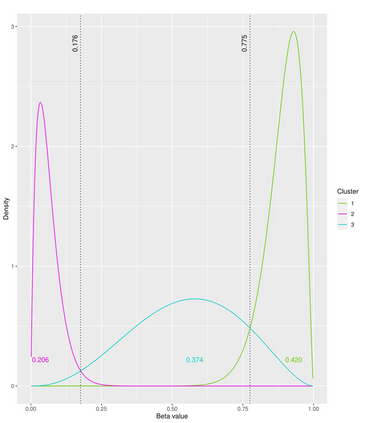
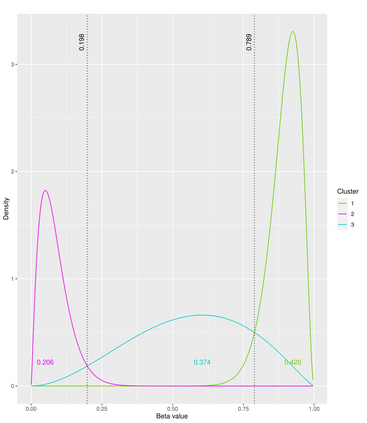
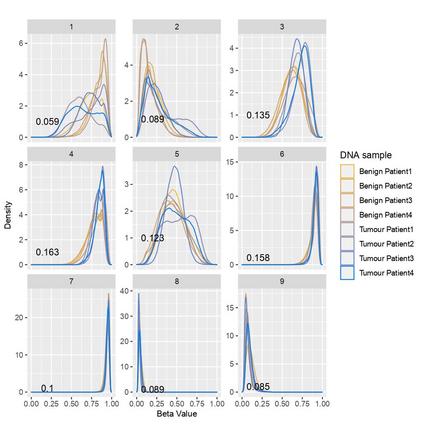
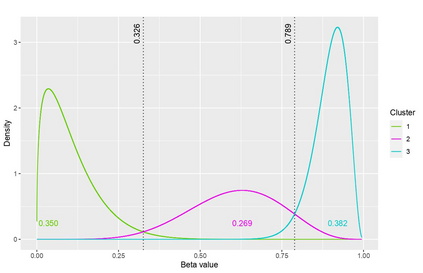
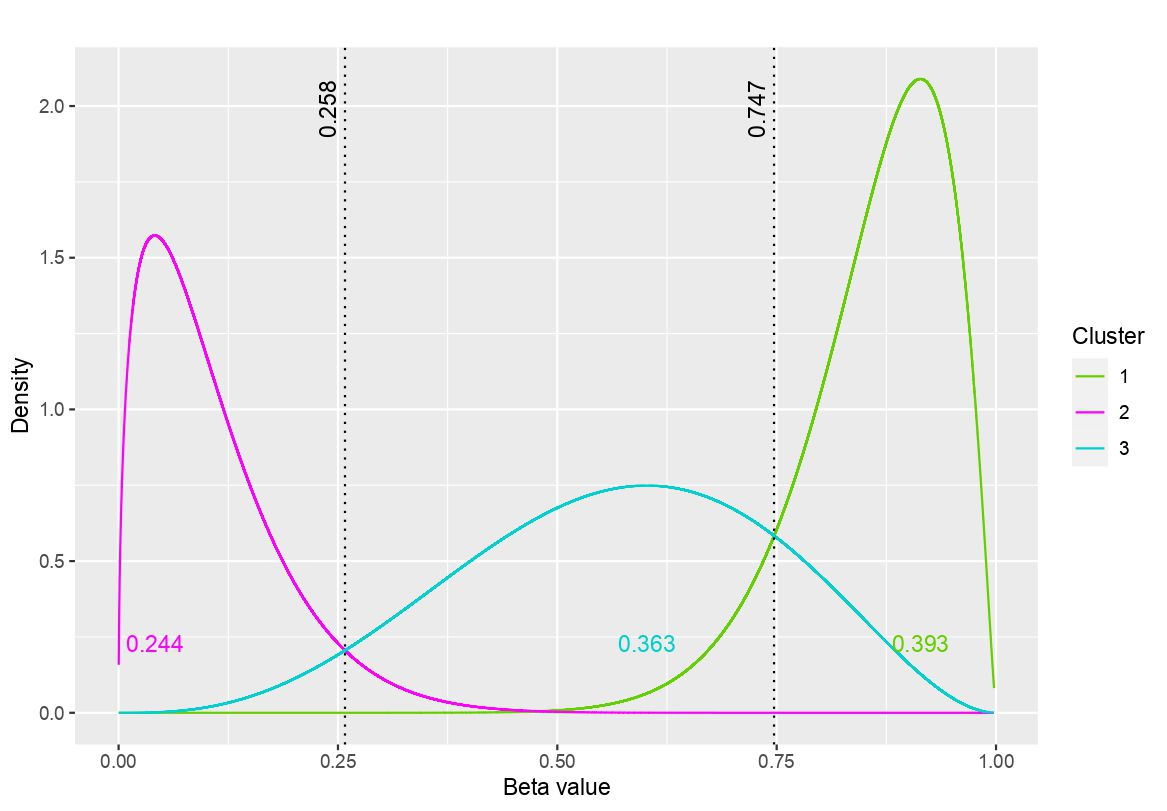
The DNA methylation process has been extensively studied for its role in cancer. Promoter cytosine-guanine dinucleotide (CpG) island hypermethylation has been shown to silence tumour suppressor genes. Identifying the differentially methylated CpG (DMC) sites between benign and tumour samples can help understand the disease. The EPIC microarray quantifies the methylation level at a CpG site as a beta value which lies within [0,1). There is a lack of suitable methods for modelling the beta values in their innate form. The DMCs are identified via multiple t-tests but this can be computationally expensive. Also, arbitrary thresholds are often selected and used to identify the methylation state of a CpG site. We propose a family of novel beta mixture models (BMMs) which use a model-based clustering approach to cluster the CpG sites in their innate beta form to (i) objectively identify methylation state thresholds and (ii) identify the DMCs between different samples. The family of BMMs employs different parameter constraints that are applicable to different study settings. Parameter estimation proceeds via an EM algorithm, with a novel approximation during the M-step providing tractability and computational feasibility. Performance of the BMMs is assessed through a thorough simulation study, and the BMMs are used to analyse a prostate cancer dataset and an esophageal squamous cell carcinoma dataset. The BMM approach objectively identifies methylation state thresholds and identifies more DMCs between the benign and tumour samples in both cancer datasets than conventional methods, in a computationally efficient manner. The empirical cumulative distribution function of the DMCs related to genes implicated in carcinogenesis indicates hypermethylation of CpG sites in the tumour samples in both cancer settings. An R package betaclust is provided to facilitate the use of the developed BMMs.
翻译:对DNA甲基化过程进行了广泛的研究,以了解其在癌症中的作用。促进细胞-夸奈二核糖酸(CpG)岛的超甲基化可以抑制肿瘤抑制基因。辨别良性与肿瘤样本之间有差异的甲基CpG(DMC)站点可以帮助理解该疾病。EPIC微观阵列将CpG站点的甲基化水平作为乙型值进行了广泛研究,该站点位于[0]之内。缺乏适当的方法来模拟产卵形式的贝塔值。DMC是通过多次测试确定的,但这种测试可能计算得非常昂贵。此外,往往选择并使用任意阈值来确定肿瘤抑制基因抑制基因的基因。我们建议使用新型乙基混合物模型来将CpG站点集中起来,以便(i)客观地确定马氏值临界值临界值,以及(ii)在不同的样品之间确定DMC。BMMC家庭使用不同的参数限制,而这些参数可以适用于不同的研究环境;此外,任意阈阈阈值确定了Cloraloral 数据在Bralalimal mal 的模型中进行数据分析。