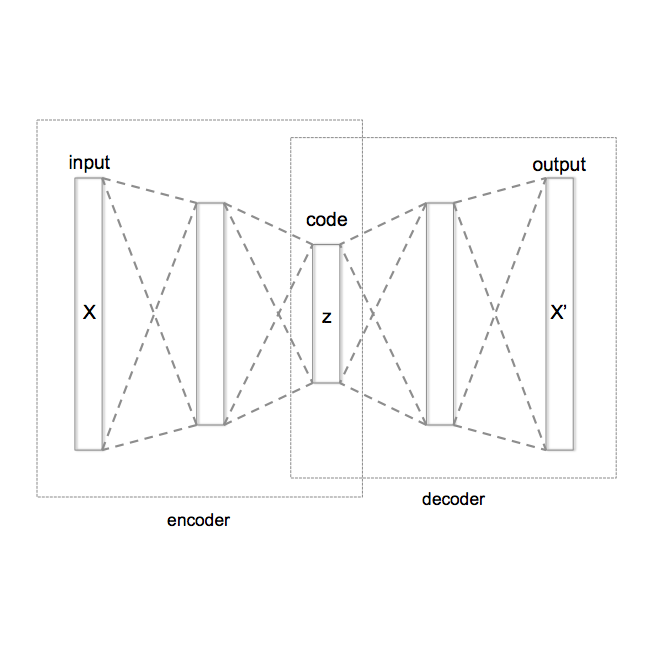
Defining and separating cancer subtypes is essential for facilitating personalized therapy modality and prognosis of patients. The definition of subtypes has been constantly recalibrated as a result of our deepened understanding. During this recalibration, researchers often rely on clustering of cancer data to provide an intuitive visual reference that could reveal the intrinsic characteristics of subtypes. The data being clustered are often omics data such as transcriptomics that have strong correlations to the underlying biological mechanism. However, while existing studies have shown promising results, they suffer from issues associated with omics data: sample scarcity and high dimensionality. As such, existing methods often impose unrealistic assumptions to extract useful features from the data while avoiding overfitting to spurious correlations. In this paper, we propose to leverage a recent strong generative model, Vector Quantized Variational AutoEncoder (VQ-VAE), to tackle the data issues and extract informative latent features that are crucial to the quality of subsequent clustering by retaining only information relevant to reconstructing the input. VQ-VAE does not impose strict assumptions and hence its latent features are better representations of the input, capable of yielding superior clustering performance with any mainstream clustering method. Extensive experiments and medical analysis on multiple datasets comprising 10 distinct cancers demonstrate the VQ-VAE clustering results can significantly and robustly improve prognosis over prevalent subtyping systems.
翻译:界定和区分癌症子类型对于便利个人化治疗模式和病人预测至关重要。子类型的定义由于我们加深理解而不断调整。在这种重新校正过程中,研究人员往往依靠癌症数据群集来提供直观的直观参考,以揭示子型的内在特征。被分组的数据往往是诸如与基本生物机制密切相关的笔录缩记式(VQ-VAE)等缩影数据。然而,虽然现有研究显示有希望的结果,但它们受到与迷宫数据有关的问题的影响:抽样稀缺和高度多维度。因此,现有方法往往强加不切实际的假设,从数据中提取有用的特征,同时避免过度适应虚假的关联。在本文件中,我们提议利用最近的强型配制模型,Vctor Qalatization Vatication Aut Encorder(VQ-VAE),以解决数据问题,并提取对随后的集成质量至关重要的信息潜在特征,只保留与重建投入有关的信息。VQV-VAE没有严格的假设,因此,现有方法往往要求从数据中提取精确的假定QQQQQQQ-因此,以更精确的模型分析。在10号上,可以更精确地分析。