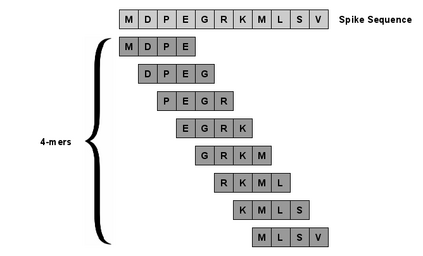
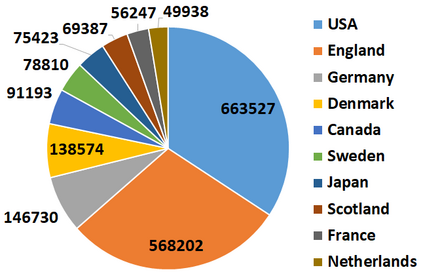
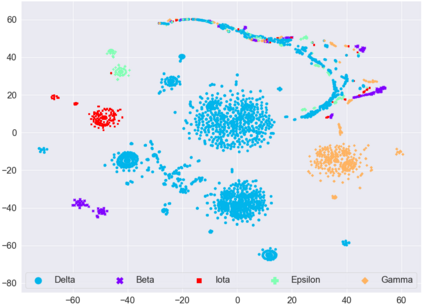
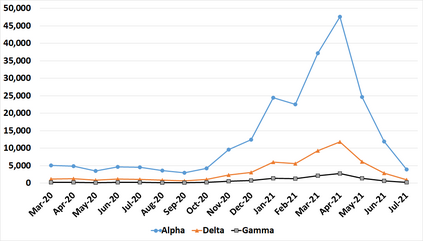
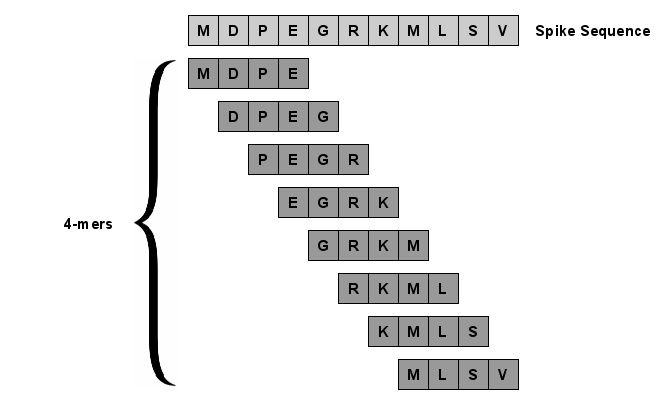
With the rapid global spread of COVID-19, more and more data related to this virus is becoming available, including genomic sequence data. The total number of genomic sequences that are publicly available on platforms such as GISAID is currently several million, and is increasing with every day. The availability of such \textit{Big Data} creates a new opportunity for researchers to study this virus in detail. This is particularly important with all of the dynamics of the COVID-19 variants which emerge and circulate. This rich data source will give us insights on the best ways to perform genomic surveillance for this and future pandemic threats, with the ultimate goal of mitigating or eliminating such threats. Analyzing and processing the several million genomic sequences is a challenging task. Although traditional methods for sequence classification are proven to be effective, they are not designed to deal with these specific types of genomic sequences. Moreover, most of the existing methods also face the issue of scalability. Previous studies which were tailored to coronavirus genomic data proposed to use spike sequences (corresponding to a subsequence of the genome), rather than using the complete genomic sequence, to perform different machine learning (ML) tasks such as classification and clustering. However, those methods suffer from scalability issues. In this paper, we propose an approach called Spike2Vec, an efficient and scalable feature vector representation for each spike sequence that can be used for downstream ML tasks. Through experiments, we show that Spike2Vec is not only scalable on several million spike sequences, but also outperforms the baseline models in terms of prediction accuracy, F1-score, etc.
翻译:随着COVID-19的迅速全球传播,越来越多的与这种病毒相关的数据正在出现,包括基因组序列数据。在GISAID等平台上公开提供的基因组序列总数目前为几百万,而且每天都在增加。这种序列分类的提供为研究人员提供了一个新的机会,以便详细研究这种病毒。随着COVID-19变种的所有动态的出现和传播,这一点尤其重要。这一丰富的数据源将使我们深入了解对这种和今后流行病威胁进行基因组监测的最佳方法,最终目标是减轻或消除这种威胁。分析和处理几百万个基因组序列是一个具有挑战性的任务。尽管传统的序列分类方法已证明是有效的,但它们并不是设计用来处理这些特定的基因序列。此外,大多数现有方法也面临着可缩缩缩问题。先前的研究仅针对可调控性病毒序列,但可调控性基因组数据只能用于直线序列(corabiting cal roadal moudalalalal modelisionalal), 而不是用来进行这种序列的不断变缩的序列, 而不是用来进行这种变序。