BMC| AI 预测多层网络组学间相互作用(药物-靶蛋白、转录因子-DNA元件和miRNA-mRNA)
22年1月26号发表在BMC上的最新文章“A deep learning approach to predict inter‑omics interactions in multi‑layer networks”提出了数据集成深度学习(DIDL)方法,预测多层网络组学间相互作用。DIDL可以在不同网络上进行评估,如药物-靶蛋白、转录因子-DNA元件和miRNA-mRNA。

开发预测组学间联系的策略对于构建疾病的整体地图至关重要,为了预测组学间的相互作用,虽然已经开发了许多用于组学数据集成的方法。然而,它们主要取决于网络节点的特定生化特性。因此,它们一般只限于特定的网络类型。
DIDL通过构建整合网络为理解复杂疾病的潜在机制铺平了道路,该算法仅依赖于已有的层间相互作用以及相互作用分子的生物化学特性,使得该算法适用于各种不同的网络。最后通过文献验证了新预测结果的有效性。
网络嵌入,是最近提出的一种方法,它通过捕捉网络的拓扑属性和边信息,将网络节点嵌入到称为潜在特征的低维向量空间中。换句话说,该方法计算成对节点之间的相似度,以找到隐藏在相应的高维数据中的低维流形结构。
基于网络嵌入的交互预测方法之一是矩阵分解,它从网络拓扑中检测潜在特征。矩阵分解数据融合(DFMF)是一种预测异构节点之间直接和间接交互的方法。
深度学习是一种从海量、异构、高维数据集的原始数据中自动提取高层特征的机器学习技术,非常适合于生物学中大数据的复杂性。将矩阵分解和深度学习相结合的思想被称为深度矩阵分解(DMF)。
该方法使用两个深度神经网络(DNN)提取表示,并作为不可训练的解码器通过余弦函数计算表示的相似度。DMF用于推荐系统,并已被证明优于传统的矩阵分解,最近已被用于预测药物与靶点的相互作用。张量分解是处理各种异构、稀疏和大数据的多层网络的有力工具。
DIDL由一个具有两个DNN的编码器和一个张量分解预测器组成,两个DNN用于提取考虑节点异质性的生物实体的表示,张量分解预测器用于预测相互作用的概率。它连接不同的层,而不依赖于相互作用分子的特定生化特性。本文在药物靶蛋白、转录因子(TF)-DNA元件和miRNA-mRNA三个不同的生物数据集上进行了评估,验证该方法的适用性。
研究方法
异质生物分子之间的相互作用是基于生物学原理的。miRNA针对的是一组在功能上相关的基因,而转录因子调控的是在其上游包含特定序列的一组基因。因此可以基于这两个节点中的每一个与相对层中的其他元素之间的已知交互来估计两个不同层中的两个给定节点之间的交互概率。
DIDL方法有两个主要组件:编码器:两个DNN在邻接矩阵上操作并产生第一和第二组学层的生物分子的隐藏特征。预测器:基于隐藏特征的张量分解模型预测交互概率。一旦编码器计算完特征向量,就设计了一个预测器应用这些特征向量来预测交互的存在,该预测器旨在计算异质生物分子之间相互作用的概率。DIDL方法框架如下(图1):
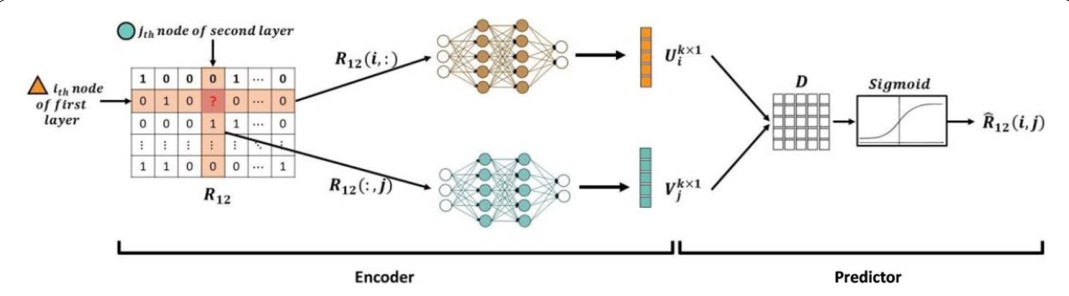
药物-靶点相互作用预测
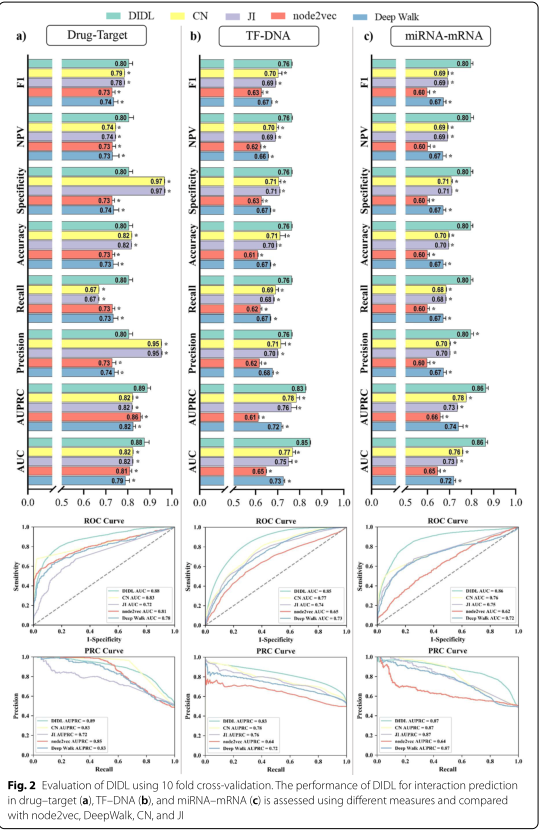
药物重新定位是药物发现中一种很有前途的方法。为了证明文中所提出的模型在这项研究中用于药物重新定位的能力,DIDL方法被用来预测药物和蛋白质之间的新联系。为此,从DrugBank数据库中提取了已知的药物-靶相互作用。这个数据集总共涵盖了1507种药物、1642种目标蛋白和6439种相互作用。为了评估DIDL在药物靶点预测中的应用,进行了10次交叉验证程序,评估了不同的指标,包括受试者工作特征曲线下面积(AUC)、精确召回曲线下面积(AUPRC)、精确度、召回率和准确度指标。通过与node2vec、DeepWalk、CN和JI的比较,经T检验分析,DIDL法优于上述三种方法(P<0.05,图2a)。
TF-DNA相互作用预测
基因表达的转录调控是转录因子与特定的DNA序列元件相互作用的结果,称为转录因子结合位点(TFBSS),是控制细胞行为的关键步骤。为了评估DIDL预测TF和TFBs之间联系的有效性,使用CHEA 2016从Enrichr数据库中提取了关于人类TF-TFBs的已知实验数据。该数据集包含总共175TF、35116个基因和407245个相互作用的数据。DIDL利用这些已知TF-DNA相互作用的数据来预测不可预见的相互作用。进行了10次交叉验证方案,并测量了性能指标。再次应用了node2vec、DeepWalk、CN和JI,并从AUC、AUPRC、查准率、召回率和准确率等方面对性能进行了评估。示,DIDL优于上述方法(P值<0.05,图2b)。
miRNA-mRNA相互作用预测
miRNA是多种细胞过程的关键调节因子,为了评估DIDL在miRNA靶预测中的性能,从miRTarBase7.0中检索了实验验证的人miRNA-mRNA相互作用,总共选择了8112个对735个miRNAs和2746个mRNAs有强有力证据的相互作用。DIDL被预测进一步的交互,最终获得了良好的性能,根据指数等于或超过0.8。T检验结果显示,DIDL法也明显优于Node2vec、DeepWalk、CN和JI(P值<0.05,图2c)。
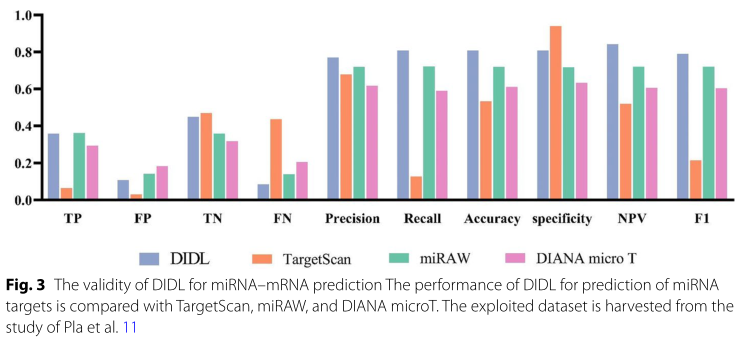
将DIDL方法与现有算法(来自Pla等人的针对miRAW数据集的TargetScan、miRAW和Diana microT的测量研究)进行比较,结果表明该方法优于其他先进的目标预测方法(图3)。
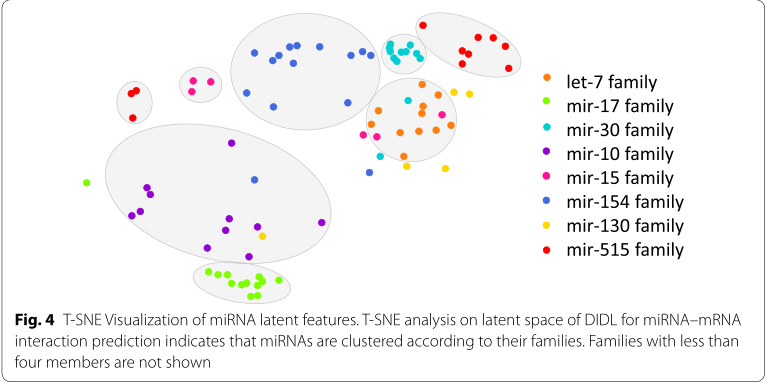
T-SNE(T-Distributed Random Neighbor Embedding)是一种非线性降维策略,其将高维空间中彼此接近的相似对象嵌入到降维空间中。作者使用T-SNE算法对编码器学习到的节点特征向量进行可视化。利用T-SNE将miRNAs的潜在特征投影到二维空间。T-SNE分析表明,miRNAs是根据它们的家族聚在一起的(图4)。在miRNA-mRNA网络中,属于同一家族的miRNAs具有相似的种子序列,因此具有相似的靶标。事实上,DIDL基于miRNA之间的相互作用以一种无监督的方式对miRNA进行聚类,这与miRNA家族是一致的。这有力地证明了该算法的有效性。
复杂多层网络中相互作用的预测
DIDL被应用于多层Hetionet网络来评估其在更复杂的上下文中用于交互预测的性能。DIDL每两层被利用一次,并且可以成功地预测这样一个复杂网络中的交互。
交互类型的预测
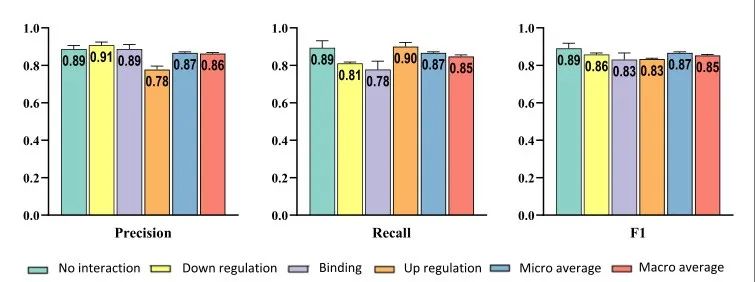
作者开发了一种可以预测交互作用类型的DIDL的修改版本,并使用Hetionet数据集的一部分对其有效性进行了评估。在这个数据集中使用了由三种不同类型的相互作用组成的基因-复合层:上调、下调和结合。DIDL不仅可以成功地预测相互作用的存在,而且可以预测它们的类型(图5)
DIDL对网络稀疏性的鲁棒性
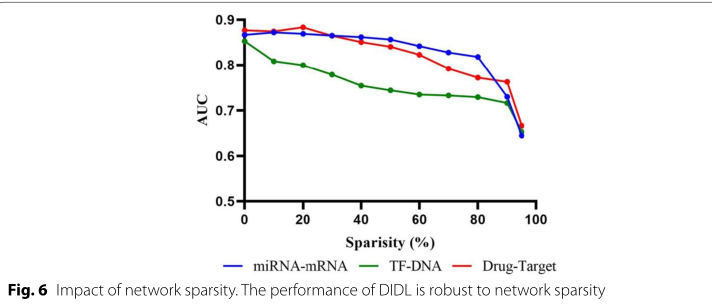
大型生物医学数据通常是高维但稀疏的。由于该方法是基于生物网络的邻接矩阵,邻接矩阵的稀疏性是影响建模性能的重要因素。因此,为了评估该方法对网络稀疏性的鲁棒性,将10%的交互作为测试子集,然后通过随机删除训练集中剩余交互的一部分来逐渐增加剩余网络的稀疏性。增加网络稀疏度会降低模型的性能。然而,模型的性能仍然可以接受,直到消除了大约50%的相互作用,特别是对于miRNA-mRNA和药物靶标数据集(图6)。
编码器效果的评估
为了研究编码器的实际影响,去掉了编码器,并将相互作用的邻接矩阵的行和列直接馈送到张量分解预测器。此修改使模型功能绝对减少。特别是对于miRNA-mRNA和药物靶向数据集,它变得近乎随机。这个实验强调了编码器对模型正确性能的重要性。
为了从整体上认识生理或病理现象的复杂机制,构建考虑异质生物分子相互作用的多层网络势在必行。本研究旨在开发一种基于深度学习的高度非线性数学数据集成方法,用于在已知相互作用的基础上预测生物网络任意两层之间的相互作用。即使在目前还不完全了解分子连接的情况下,DIDL也可以被可靠地利用。该方法的另一个优点是特征选择和网络表示过程是自动的。
综上所述,本文采用深度学习策略,提出了一种新的基于最小数据的、适用于各种网络的互组学预测流水线。它可以被用来构建多层网络,并生成复杂疾病潜在机制的全面地图。
参考文献
[1] A deep learning approach to predict inter-omics interactions in multi-layer networks