几何图神经网络在百度生物计算平台的应用


导读:本次分享的主题是几何图神经网络在药物发现中的应用。主要包括以下几部分内容:
百度生物计算平台简介
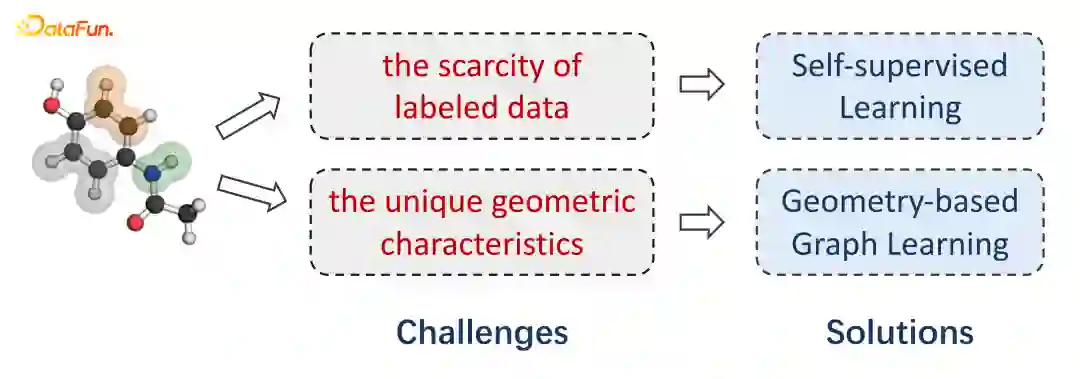
基于3D空间结构的药物亲和力预测
基于几何图神经网络的小分子性质预测
分享嘉宾|周景博博士 百度研究院 资深研究员
编辑整理|王龙飞
出品平台|DataFunTalk
百度生物平台简介

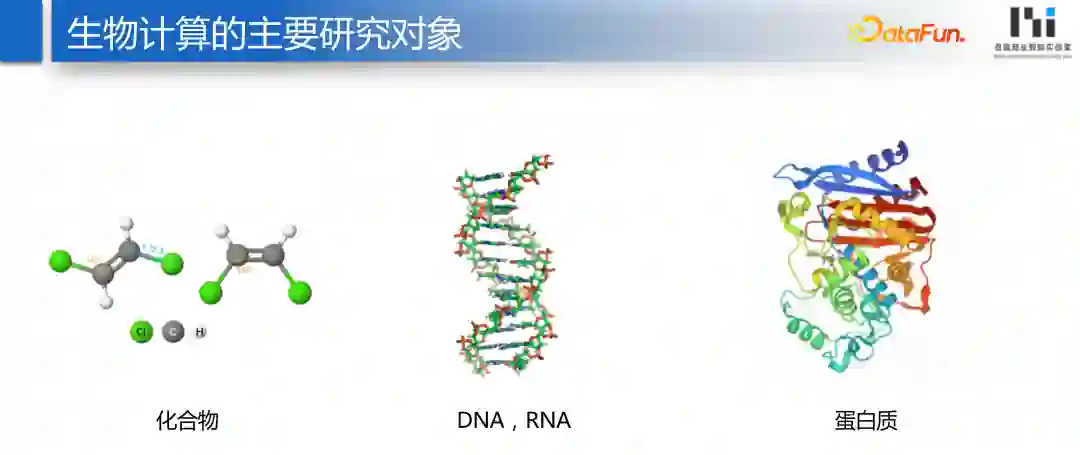
-
化合物
-
DNA、RNA
-
蛋白质

-
虚拟筛选

-
Protein-Ligand Binding Affinity
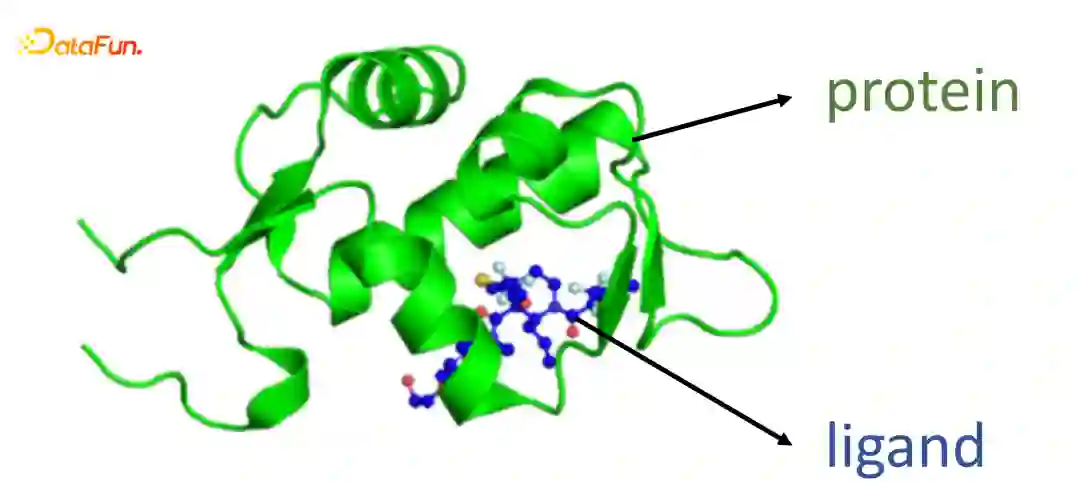
-
Structure-based Binding Affinity Prediction
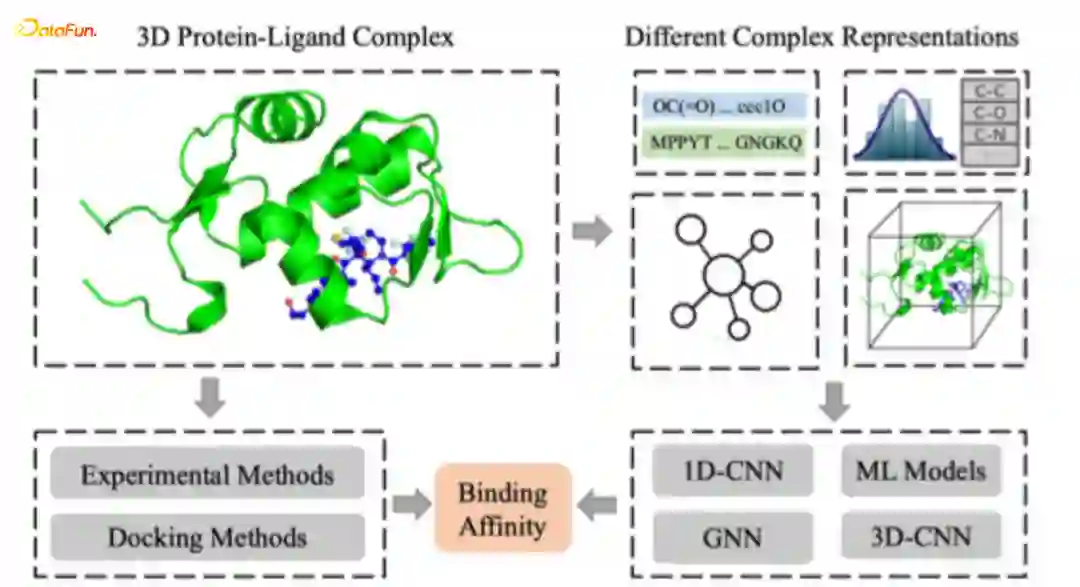
-
Complex Interaction Graph Construction
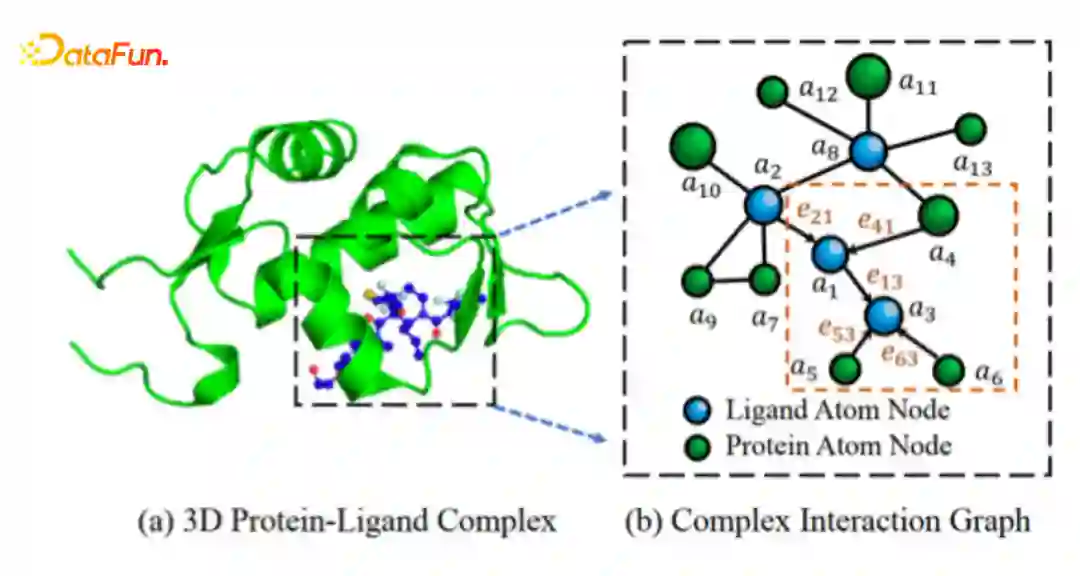
-
Structure-aware Interactive Graph Neural Network (SIGN)
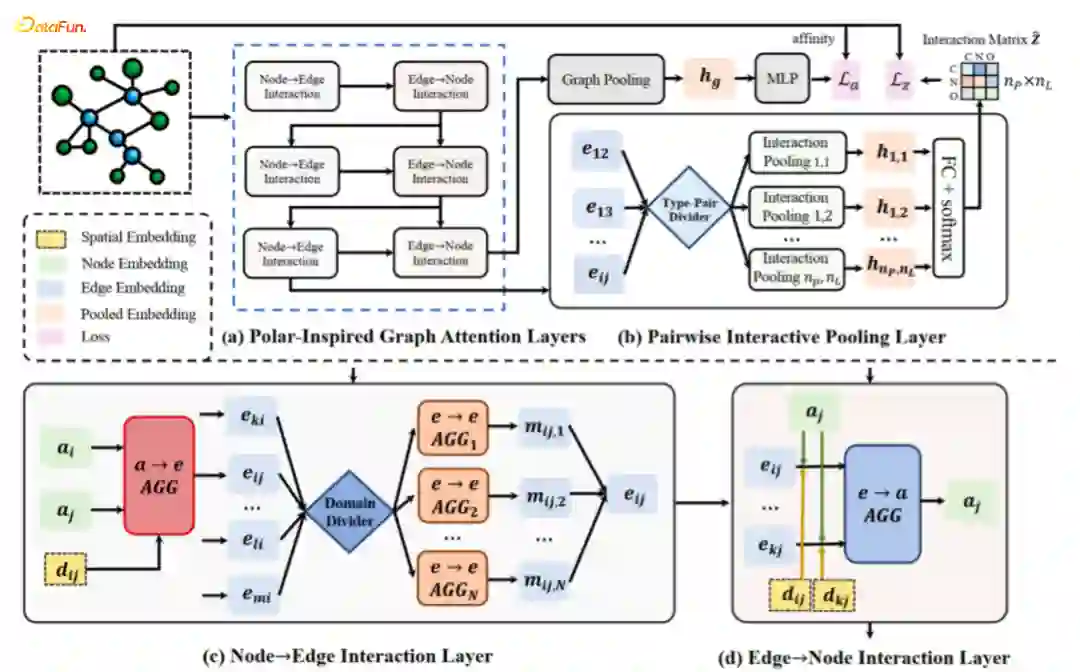
-
Polar Coordinate-Inspired Graph Attention

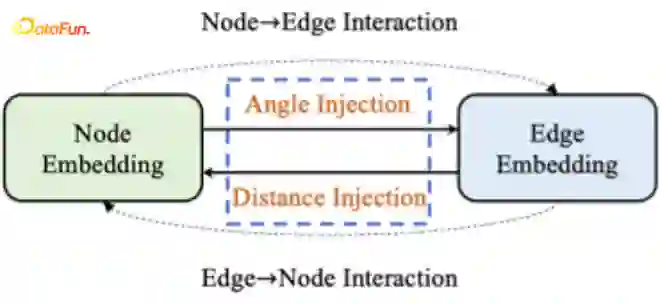

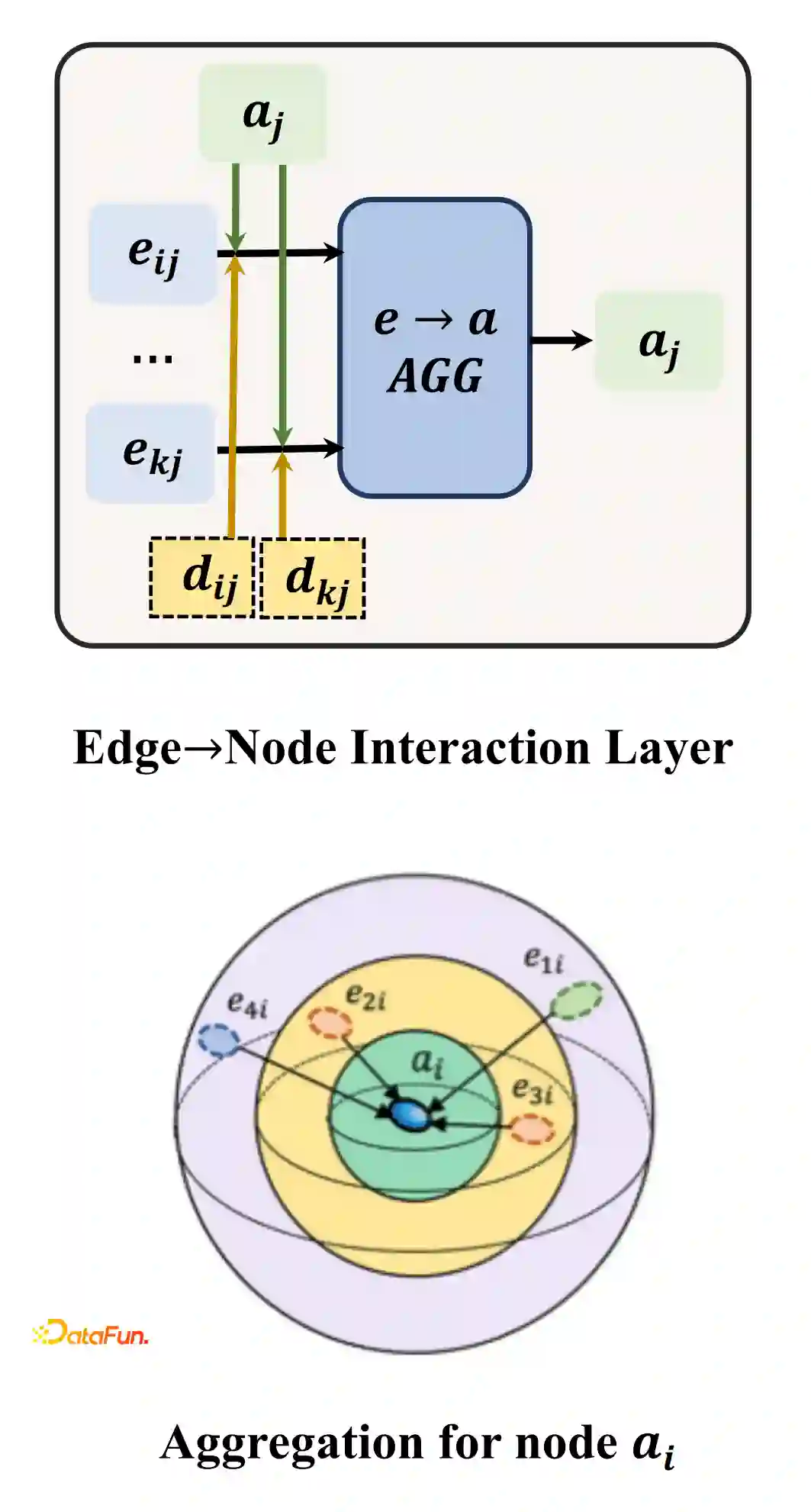
-
Datasets

-
Baselines

-
Comparison with baselines
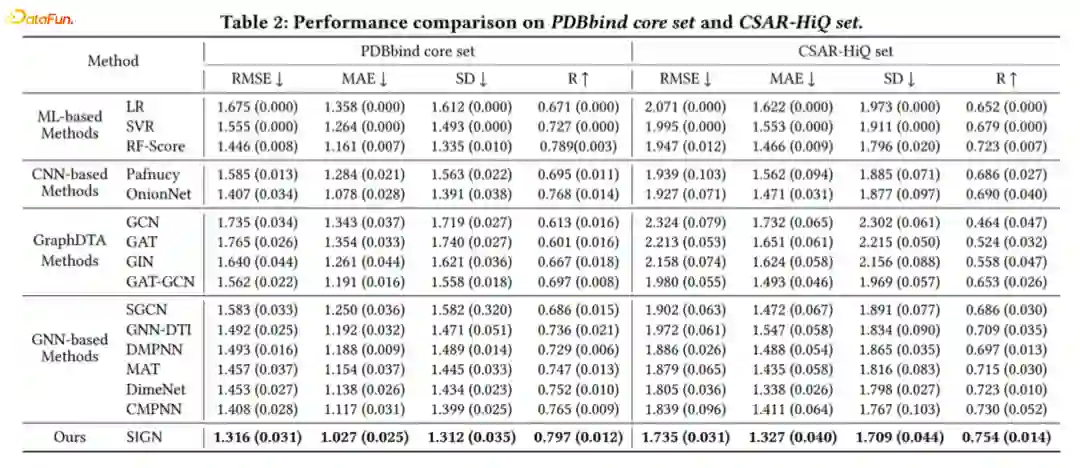
-
Impact of Spatial and Interactive Factors

-
Molecular Property Prediction

-
Graph Representation Learning for Molecules
-
Geometrics Structure Learning on Graphs
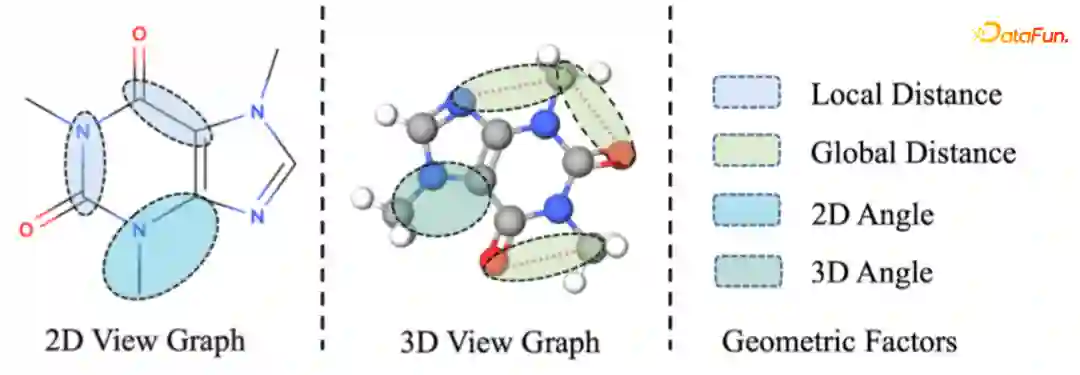
-
Contrastive Learning on Graphs

-
Geometric Graph Contrastive Learning

-
Overall Framework for GeomGCL
-
Geometry-based RBF Encoding
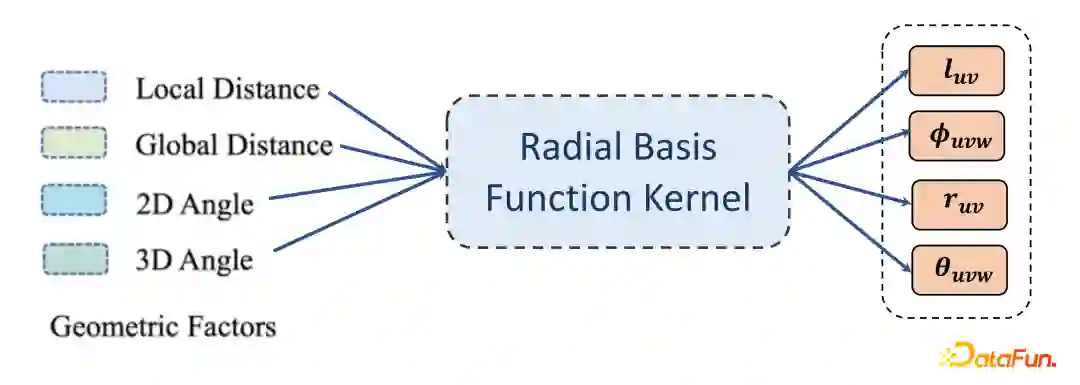
-
Adaptive Geometric Message Passing Scheme

-
Dataset

-
Baselines

-
Comparison with baselines
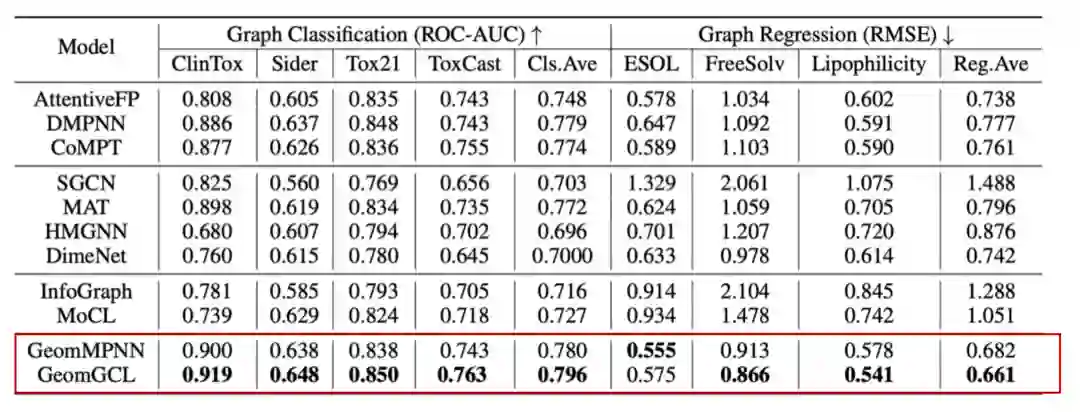

-
Summary of our work
-
化合物表征模型GEM


|分享嘉宾|

周景博 博士
百度研究院 资深研究员
周景博,现任百度研究院商业智能实验室资深研究员,主要从事数据挖掘和机器学习相关的研究和应用工作, 包括时空数据挖掘、深度几何学习和知识图谱等。2014年从新加坡国立大学获得博士学位,并于2015年加入百度研究院。他目前已经有超过30余篇论文发表在计算机顶级会议和期刊上,包括KDD, SIGMOD, ICDE, AAAI, TKDE和Lancet Public Health,Nature Machine Intelligence等,并常年担任KDD, AAAI, IJCAI, ACL, CIKM, TKDE, VLDBJ等顶级学术会议和期刊的程序委员会委员和审稿人。他作为组委会核心负责人之一承办了KDDCup 2022机器学习竞赛并担任出题人。
专知便捷查看
便捷下载,请关注专知公众号(点击上方蓝色专知关注)
后台回复“几何深度学习” 就可以获取《 几何深度学习专知资料合集》专知下载链接


登录查看更多
相关内容
Arxiv
14+阅读 · 2019年8月12日

相关VIP内容
相关资讯