没有相应的辅助因子,许多蛋白质在自然界中就不会出现。比如肌红蛋白或血红蛋白需要血红素才能发生折叠。 因此,分析蛋白质与辅助因子、配体和离子的接触,有助于完整的了解蛋白质的功能和结构。 2022年11月24日,荷兰癌症研究所的Anastassis Perrakis等人在Nat Method发表文章,提出了一种利用序列和结构相似性、将这种”缺失”的小分子和离子从实验确定的结构中”移植”到蛋白质预测模型中的算法AlphaFill,该算法在实验结构上成功验证。

摘要
基于人工智能的蛋白质结构预测方法已经对生物分子科学产生了变革性影响。然而,基于AlphaFold蛋白质结构数据库的蛋白质结构预测都缺乏结构或功能所必需的小分子配体信息**,**如血红蛋白缺乏结合的血红素,锌指 (zinc-finger) 结构缺乏结构完整性所必需的锌离子,金属蛋白酶缺乏催化所需的金属离子,对生物功能重要的配体也不存在,没有ADP或ATP与任何ATP酶或激酶结合。 本文提出了AlphaFill,一种利用序列和结构相似性将这种”缺失”的小分子和离子从实验确定的结构中”移植”到蛋白质预测模型中的算法。该算法针对实验结构成功地进行了验证。在995,411个AlphaFold模型上共进行了12,029,789次移植,并在alphafill.eu数据库中提供了相关的验证指标,这是一个帮助科学家提出新假设和设计目标实验的资源。 主要内容
AlphaFold和RoseTTAfold使蛋白质结构预测变得空前可靠。这两种方法都能以令人印象深刻的准确度预测结构域,但蛋白质的灵活部分(如环路或内在无序区)的预测准确度和置信度较低,这是可以理解的。 人工智能预测算法并没有被训练成从第一性原理解决蛋白质折叠问题。它们只是在对实验解决的结构进行广泛训练的基础上,学习了蛋白质折叠的固有规则。
然而,许多蛋白质在自然界中没有它们的辅助因子就不会出现:肌红蛋白或血红蛋白需要血红素才能折叠,锌指结构域没有锌离子就不稳定。AlphaFoldMultimer和RoseTTAFold的开发解决了多聚体的问题,它们可以预测复杂的蛋白质组合。然而,预测的结构模型只考虑了20个典型的氨基酸残基,并没有预测通常与蛋白质相关的小分子、配体和辅助因子的情况。 本文通过”移植”在同源蛋白质结构中实验观察到的小分子和离子的信息来丰富AlphaFold数据库中的模型。所提出的AlphaFill程序已经针对实验结构进行了验证,并应用于所有AlphaFold模型,以创建一个新的资源,即AlphaFill数据库,旨在帮助生命科学家轻松产生新的蛋白质功能假设,并制定相关的研究问题。 结果
移植化合物到AlphaFold模型
首先,作者在PDB-REDO数据库的AlphaFold数据库中为每个结构搜索序列同源物。 接下来,在AlphaFold模型的Cα原子上对所选结构与感兴趣的化合物进行结构比对,并计算出均方根偏差(r.m.s.d) (全局r.m.s.d)。从最接近的同源物开始,选择距离每个将被考虑”移植”的化合物的原子6 Å范围内的所有骨干原子,并用于与AlphaFold模型进行局部结构比对;同时计算该比对的均方根偏差 (r.m.s.d.)。然后将化合物移植到AlphaFold模型中,形成AlphaFill模型。
AlphaFill数据库
将AlphaFill方法应用于2022年2月的AlphaFold数据库(995,411个模型),结果有586,137个模型至少有一个移植的化合物。总共有12,029,789个化合物被移植到这些模型中。
AlphaFill算法的验证
为了验证AlphaFill算法,作者将AlphaFill创建的移植体与具有100%序列一致性的实验结构进行了比较。将局部环境验证(LEV)得分定义为AlphaFill和实验复合物之间的任何配体原子和距离配体6.0 Å内的所有蛋白质原子的全原子密度。图1a显示了该验证集(28619个移植体)中所有AlphaFill结构的LEV得分分布。
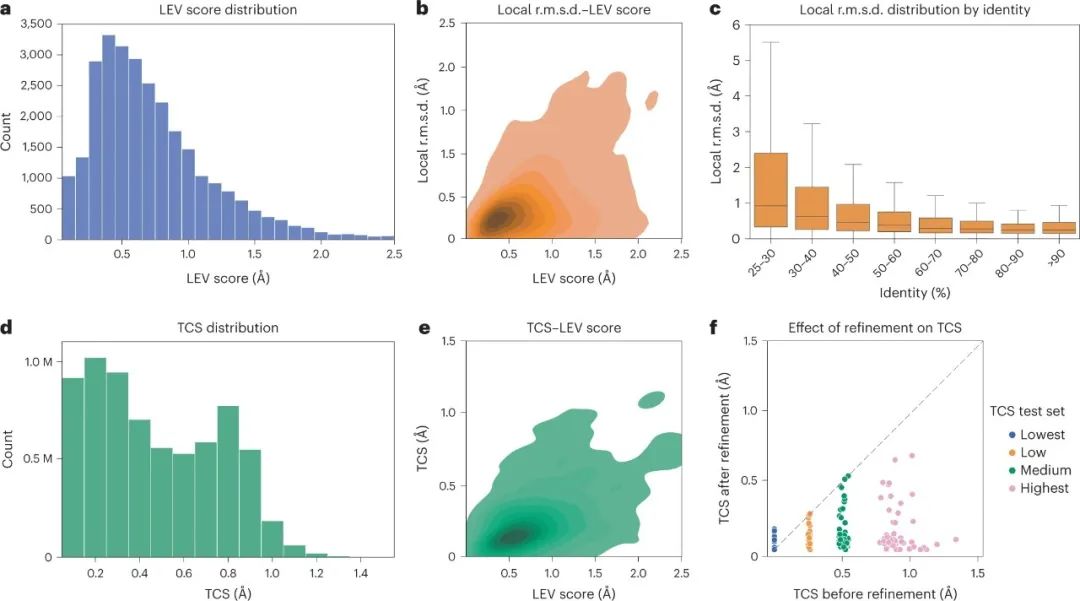
图1:AlphaFill算法的验证 验证移植质量的一个正交方法是评估配体和蛋白质原子之间可能发生的冲突。为此,作者将移植冲突得分(TCS)定义为移植配体与其结合部位之间的范德瓦尔斯重叠的函数(详见方法)。图1d显示了所有多原子移植的TCS的分布。TCS与LEV分数有很好的相关性(图1e)。
对AlphaFill数据库移植的质量分析
然后,验证结果被用来得出质量指标,以注释AlphaFill数据库中的移植。由于本地r.m.s.d.与LEV评分有很好的相关性(图1b),作者进一步分析了它作为序列身份的函数的分布(图1c)来注释移植。
基于网络的AlphaFill数据库的用户界面
所有AlphaFill的条目都可以通过AlphaFill网站(https://alphafill.eu)进行视觉检查。在主页上,可以使用AlphaFold标识符检索模型,该标识符相当于UniProt的主要加入代码。 在每个条目页面上(图2),所选择的AlphaFill模型会用可视化软件Mol*来显示,允许用户完全灵活地进行检查。

图2:细胞视黄酸结合蛋白2(AF-P29373)的AlphaFill条目页面的截图
案例
对于在PDB中具有相同结构的模型,AlphaFill数据库部分复制了PDBe-Knowledge Base中已有的信息。AlphaFill也可以从同源的实验结构中移植化合物,这些实验结构可能是在另一个物种中确定的,也可以移植到有类似结构域的实验上。 作者讨论了几个例子。
肌红蛋白和血红素
人类肌红蛋白是一个ɑ-螺旋形的蛋白质,以血红素B为辅助因子,与分子氧和其他一些小分子结合。AlphaFold模型(AF-P02144)与实验确定的结构几乎相同,并显示了一个血红素形状的空腔(图3)。在AlphaFill数据库中,许多血红素类似物(含有铁以外的金属)根据CoFactor数据库中的数据被”映射”回血红素B(HEM,PDB命名法)。 缺乏羧基尾部的血红素类似物6HE和7HE没有被映射回血红素B,而是被原样移植。其他被移植到AlphaFold肌红蛋白模型的化合物包括分子氧和一氧化碳。后者被安装在两个位置:一个靠近血红素中的铁原子,另一个在血红素的远侧。第二个一氧化碳,位于一个意想不到的位置,继承自PDB-REDO条目1dwt,其中它的模型占用率为30%。**在AlphaFill模型中保留了这一占用率,以使用户在评估该模型时能考虑到这一点。**AlphaFill模型的肌红蛋白还包含许多金属离子。
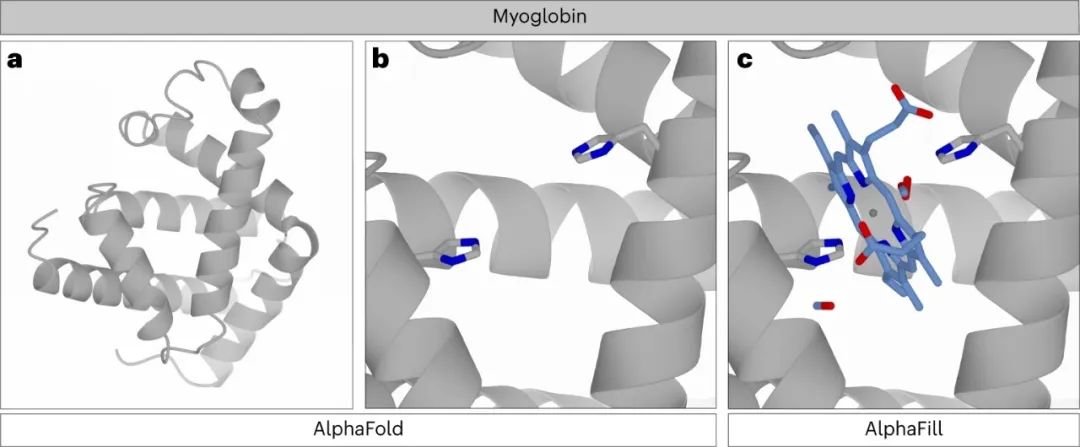
图3:AlphaFold和AlphaFill中的人类肌红蛋白结构。
a,人类肌红蛋白AlphaFold模型的带状图。b, AlphaFold模型中的血红素形状的空腔,其中组氨酸侧链(按原子类型着色的灰色圆柱体)已经准备好促进血红素的结合。c,AlphaFill模型中的血红素形状的空腔,其中结合部位被移植的血红素组和CO及O2配体”填充"。 配体按原子类型(血红素)着色的棒状模式显示,血红素铁为灰色球形。
锌的结合点
大分子结构中最常见的过渡金属离子是锌。通常,它参与催化作用或维持结构的完整性。所谓的”结构性锌离子”通常涉及一个四面体的结合点,包含四个协调的半胱氨酸和/或组氨酸残基的组合。 含有功能性和结构性锌离子的蛋白质之一是STAM结合蛋白,它是一种锌金属蛋白酶,可以裂解赖氨酸-63连接的聚泛素链(AF-O95630)。锌离子被移植到AlphaFill模型中,在催化部位和锌指图案处(图4a)。结构中的锌离子由三个组氨酸残基和一个半胱氨酸配合。虽然这个四面体的锌结合点看起来很合适,但是锌原子和其配体之间的原子距离偏离了以前确定的目标值。**这种限制是AlphaFold在关键结构元素之外预测结构的结果,在这种情况下就是锌离子。****通过添加锌原子,提供了定性的信息(锌原子应该在这个结合点)。**但是不应该从AlphaFill模型中提取关于锌结合点的定量信息。可以用几何约束条件进一步完善AlphaFill模型,使结合点看起来更正常。

图4:移植的锌离子的例子(紫色球体)。
a, STAM结合蛋白中的一个催化型(顶部)和一个结构型(底部)锌离子。b, 人类BMI-1中的两个结构性锌离子。c, 锌指蛋白91中转移到结构性锌结合位点的锌离子(上),同一蛋白中错误放置的锌离子(下)。d, 在PDB-REDO模型中发现的ENPP1-7的双金属锌结合位点,与AlphaFold的人类ENPP1-7模型和AlphaFill中的相同结合位点相比,含有两个锌离子。 在外核苷酸焦磷酸酶/磷酸二酯酶(ENPP)家族的蛋白质中,双金属锌位点对催化作用非常重要。ENPP1-7的PDB-REDO模型的催化域的结构排列(图4d)显示,锌原子和协调它们的残基在所有家族成员中占据了高度相似的位置。相同蛋白质的AlphaFold预测(ENPP1-7的AF-P22413、AF-Q13822、AF-O14638、AF-Q9Y6X5、AF-Q9UJA9、AF-Q6UWR7、AF-Q6UWV6,分别)显示出更多的分歧,特别是组氨酸R5(图4d)。AlphaFill捕捉到AlphaFold和PDB-REDO模型之间的相似性,将这两个锌离子移植到ENPPs的蛋白质模型中(图4d)。组氨酸R5在AlphaFold预测中具有不同的旋转体,根据实验结构,它应该是一个单一的旋转体,这表明AlphaFill模型中的双金属锌位点可以从额外的细化中受益。
激酶和ATP
众所周知,激酶有多种状态,一种是提供有利于磷转移反应的环境的活性构象,另一种是不满足催化活性所需的化学限制的非活性状态。**到目前为止,AlphaFold只为每个蛋白质提供一种构象。**AlphaFold模型所对应的状态,事先是不知道的。 然而,AlphaFill可以将ADP和ATP(或其类似物)转移到AlphaFold模型中,只要PDB-REDO数据库中有相关的实验结构,不管特定残基的构象所描述的激酶的功能状态如何。
对于人类酪氨酸蛋白激酶ABL1(AF-P00519),AlphaFill模型显示了一个ADP分子和一个ATP分子(图5a,b),允许对该模型的功能状态提出不同的假设。

图5:AlphaFill有助于理解Abl激酶AlphaFold模型的激活状态。
a,ABL1激酶的AlphaFill模型,显示了ADP和镁离子。b,ABL1激酶与ATP(由AGS映射)结合的AlphaFill模型。c,PDB-REDO条目2g2i(参考文献30)中人类ABL1激酶的ADP结合位点,代表活性激酶状态。 讨论
分析蛋白质与辅助因子、配体和离子的接触,有助于了解蛋白质的功能和结构完整性。它们也有助于设计下游实验,无论是计算还是在湿法实验室。到目前为止,AlphaFold数据库不包括这些化合物。 AlphaFill软件是免费提供的(在BSD许可下),它允许用户”提交”任何结构模型进行评估,也有可能考虑PDB中所有>30,000个非聚合物配体。一个API允许用户上传和”填充”他们自己的模型或AlphaFold数据库中的额外结构(2022年6月后添加),也提供了访问PDB中额外非聚合物化合物的机会。目前AlphaFill并不处理聚合物配体,如肽、核酸或糖类。它也不处理翻译后修饰,特别是糖基化。其他翻译后的修饰,如磷酸化,经常会引起构象的变化,AlphaFill也同样没有处理。 AlphaFill结构模型并不意味着是准确的、精确的或完整的代表某种蛋白质结构的全部配体。它们是作为非专业人员的工具,帮助他们探索常见配体的复合体。 需要注意的是,AlphaFill模型并不适合精确量化转移配体和蛋白质之间的相互作用(例如,氢键、π-π或阳离子-π相互作用、范德瓦尔斯相互作用、疏水相互作用、卤素键)。也就是说,这需要AlphaFold或AlphaFill模型所不能提供的配体情况的精度(即使经过优化)。因此,应该以定性的方式来解释这些模型。此外,在某些情况下,配体的相互作用涉及到蛋白质的某些部分,而AlphaFold的模型并不具有很高的可信度;虽然优化可能会改善局部环境,但我们建议要谨慎。 根据定义,AlphaFill依赖于高质量的结构同源物作为转移配体的第一和主要标准。然而,正如DALI和PDBeFold所显示的那样,某些结构域可以在广泛的序列相似性之外出现,这一点已得到公认。因此,AlphaFill可以通过基于深度学习概念的基于结构的转移算法进行补充,类似于用于AlphaFold结构预测革命的算法。
参考资料 Hekkelman, M.L., de Vries, I., Joosten, R.P. et al. AlphaFill: enriching AlphaFold models with ligands and cofactors. Nat Methods (2022). https://doi.org/10.1038/s41592-022-01685-y
--------- End ---------