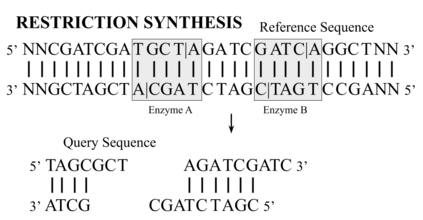
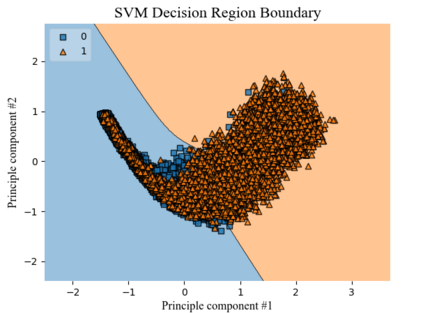
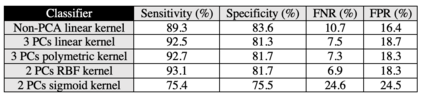
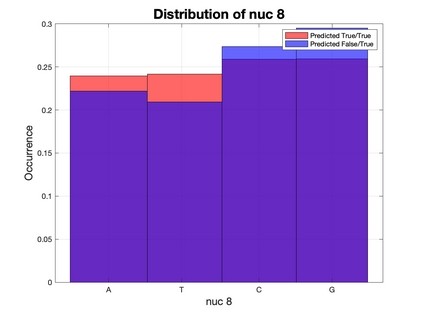
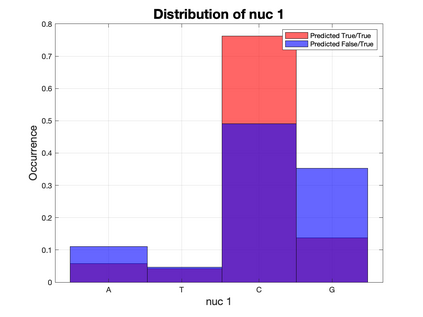
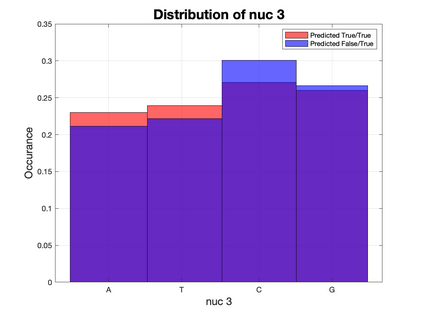
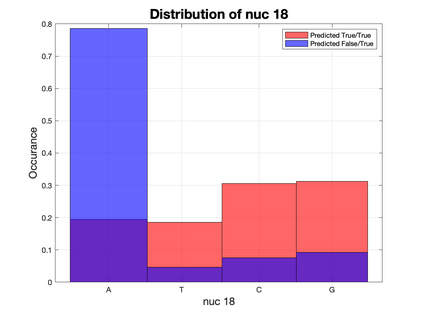
Based on the BioBricks standard, restriction synthesis is a novel catabolic iterative DNA synthesis method that utilizes endonucleases to synthesize a query sequence from a reference sequence. In this work, the reference sequence is built from shorter subsequences by classifying them as applicable or inapplicable for the synthesis method using three different machine learning methods: Support Vector Machines (SVMs), random forest, and Convolution Neural Networks (CNNs). Before applying these methods to the data, a series of feature selection, curation, and reduction steps are applied to create an accurate and representative feature space. Following these preprocessing steps, three different pipelines are proposed to classify subsequences based on their nucleotide sequence and other relevant features corresponding to the restriction sites of over 200 endonucleases. The sensitivity using SVMs, random forest, and CNNs are 94.9%, 92.7%, 91.4%, respectively. Moreover, each method scores lower in specificity with SVMs, random forest, and CNNs resulting in 77.4%, 85.7%, and 82.4%, respectively. In addition to analyzing these results, the misclassifications in SVMs and CNNs are investigated. Across these two models, different features with a derived nucleotide specificity visually contribute more to classification compared to other features. This observation is an important factor when considering new nucleotide sensitivity features for future studies.
翻译:根据BioBricks标准,限制合成是一种新颖的代谢迭代迭代DNA合成方法,它利用内分泌来合成一个参考序列中的查询序列。在这项工作中,参考序列是从较短的子序列中建立的,方法是使用三种不同的机器学习方法,将它们分类为可适用或不适用于合成方法:支持矢量机(SVMS)、随机森林和进化神经网络(CNNs),在对数据应用这些方法之前,采用了一系列特征选择、调整和减少步骤来创建准确和有代表性的特征空间。在这些预处理步骤之后,建议三个不同的管道根据核核酸序列和与200多个内分泌排放限制地点相对应的其他相关特征对次序列进行分类。使用SVMS、随机森林和CNNN(CNs)的敏感性分别为94.9%、92.7%、91.4%。此外,每种方法在与SVM、随机森林和CNN等新特性的特性下分数较低,结果为77.4%、85.7%和82.4%。此外,在对S-MICLCL的特性进行这些不同分析时,这些结果将分别纳入。