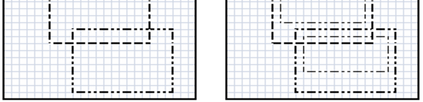
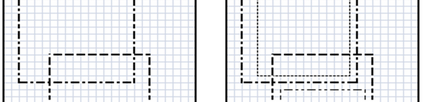
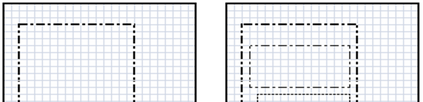
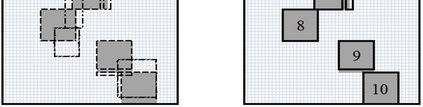
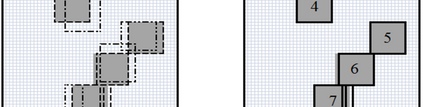
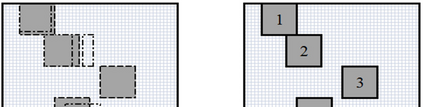
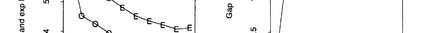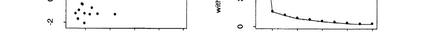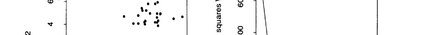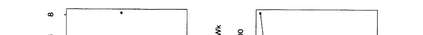The availability of large microarray data has led to a growing interest in biclustering methods in the past decade. Several algorithms have been proposed to identify subsets of genes and conditions according to different similarity measures and under varying constraints. In this paper we focus on the exclusive row biclustering problem for gene expression data sets, in which each row can only be a member of a single bicluster while columns can participate in multiple ones. This type of biclustering may be adequate, for example, for clustering groups of cancer patients where each patient (row) is expected to be carrying only a single type of cancer, while each cancer type is associated with multiple (and possibly overlapping) genes (columns). We present a novel method to identify these exclusive row biclusters through a combination of existing biclustering algorithms and combinatorial auction techniques. We devise an approach for tuning the threshold for our algorithm based on comparison to a null model in the spirit of the Gap statistic approach. We demonstrate our approach on both synthetic and real-world gene expression data and show its power in identifying large span non-overlapping rows sub matrices, while considering their unique nature. The Gap statistic approach succeeds in identifying appropriate thresholds in all our examples.
翻译:在过去十年中,大量微观阵列数据的可用性促使人们对双组制方法的兴趣日益浓厚。提出了几种算法,以便根据不同的类似措施和各种限制,确定基因表达数据集的基因和条件子集。在本文件中,我们侧重于基因表达数据集的单行双组问题,即每行只能是单一双组组的成员,而各纵队可以参加多个组。这种双组制可能足够,例如,对于每个病人(row)只携带单一类型癌症的癌症患者组群,而每种癌症类型都与多种(可能重叠的)基因(栏目)有关。我们提出了一个新颖的方法,通过将现有的双组式算法和组合拍卖技术结合起来,来识别这些独列双组。我们设计了一种方法,根据对差距统计方法精神的无效模型进行比较,调整我们的算法门槛。我们展示了我们关于合成和真实世界基因表达数据的方法,并展示了它在确定大跨非重叠行子矩阵(栏)方面的力量,同时考虑它们的独特性。我们的所有模型都成功地确定了我们的标准。