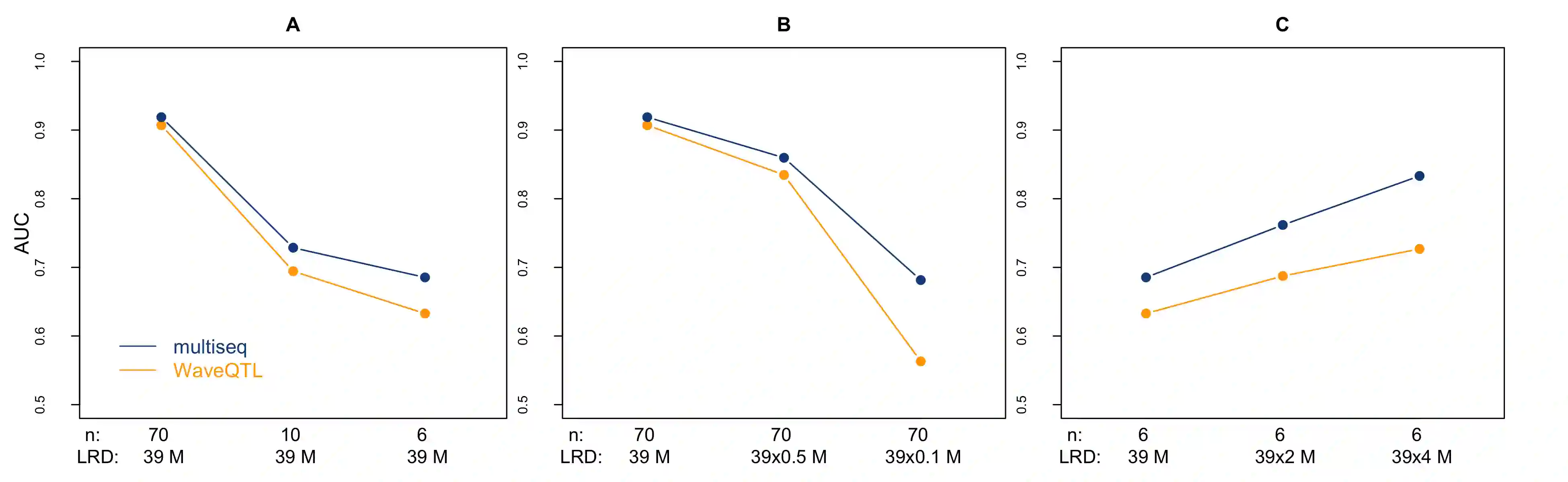
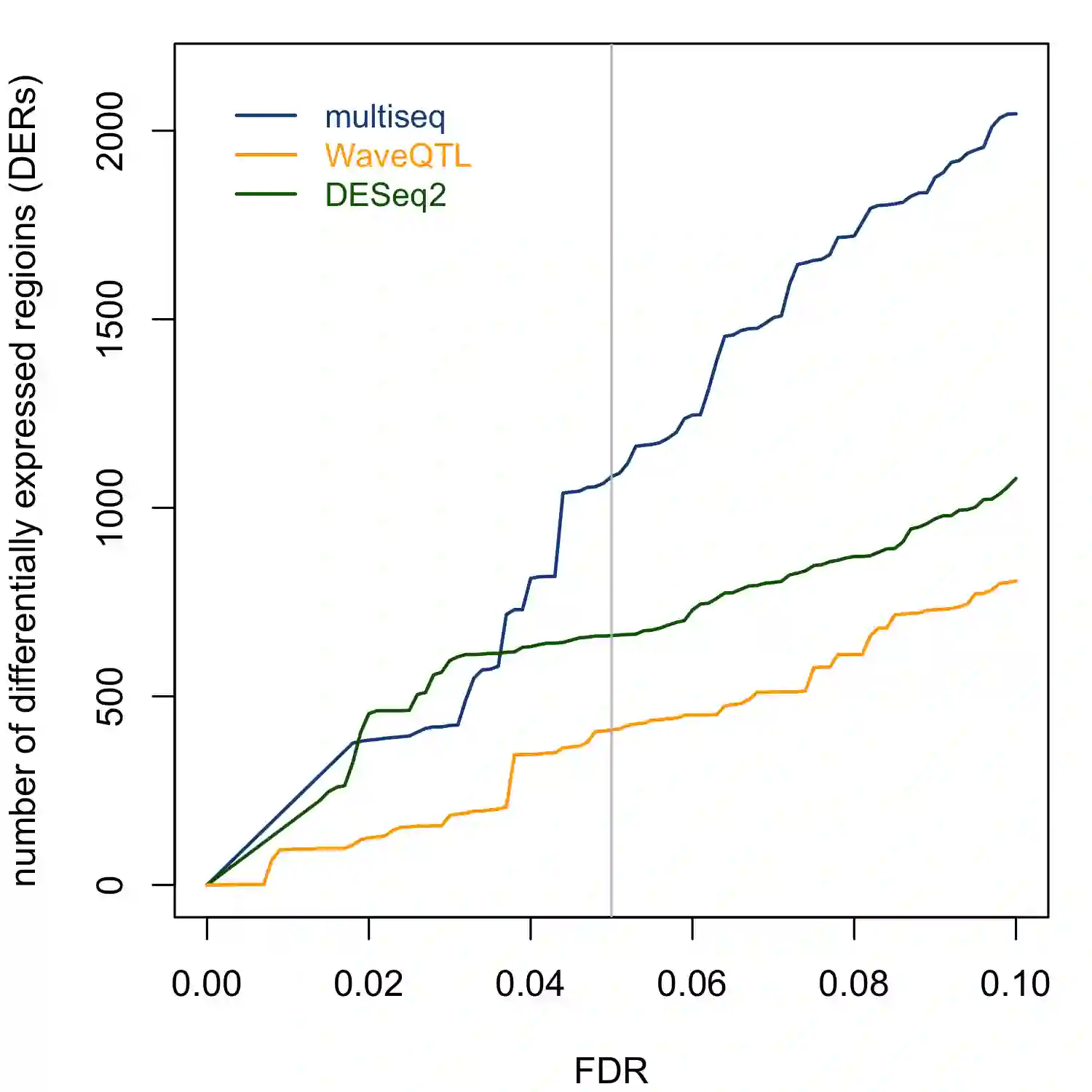
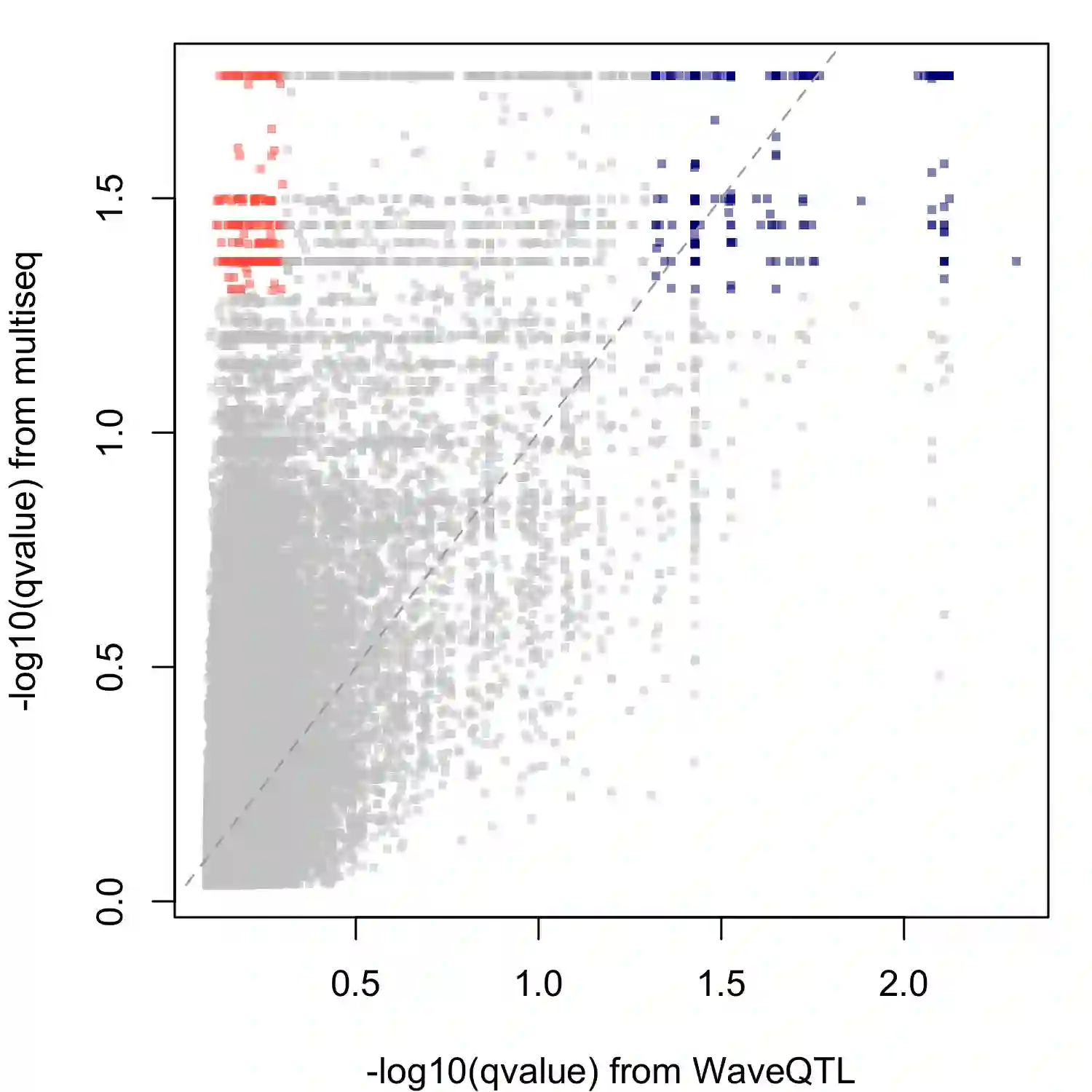
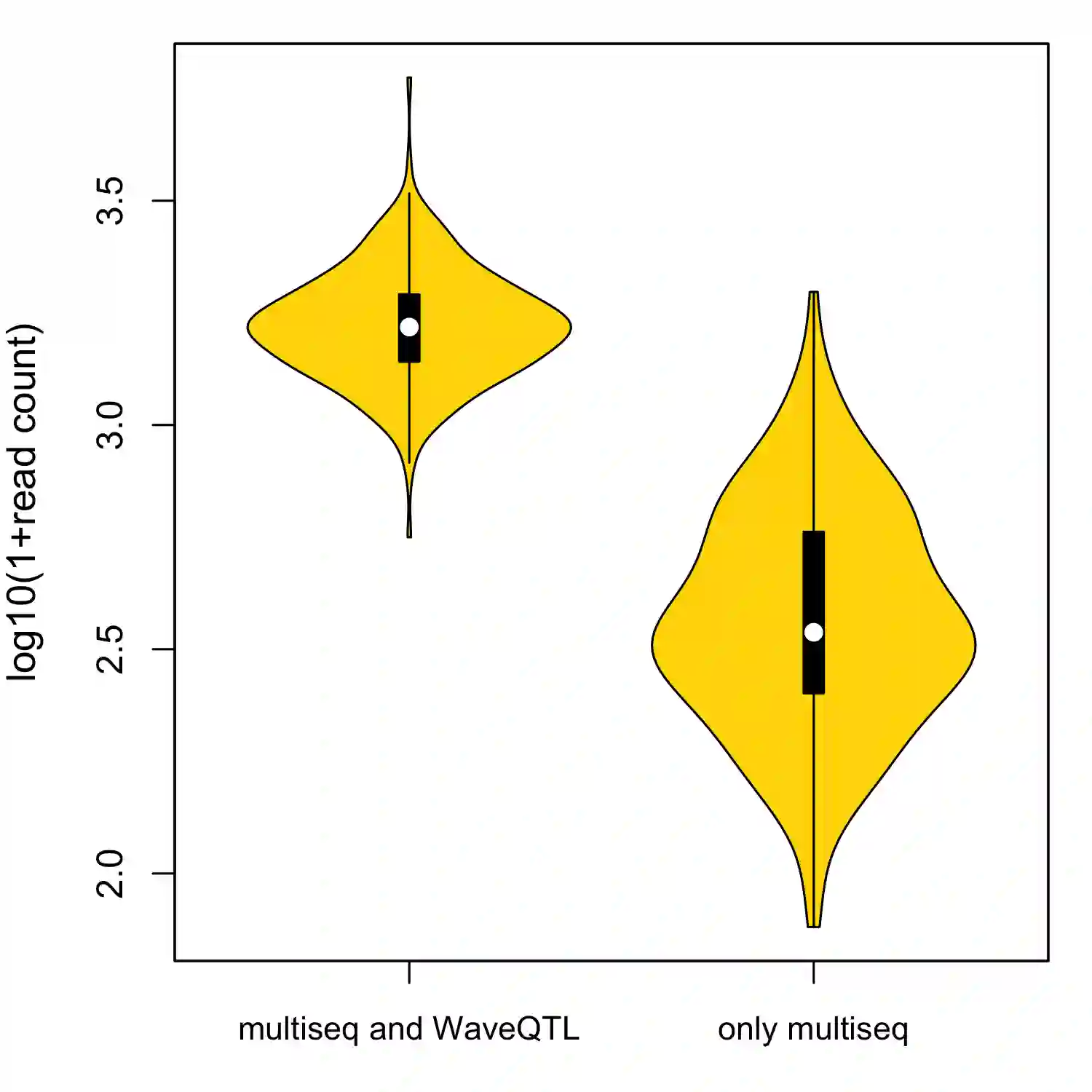
Estimating and testing for differences in molecular phenotypes (e.g. gene expression, chromatin accessibility, transcription factor binding) across conditions is an important part of understanding the molecular basis of gene regulation. These phenotypes are commonly measured using high-throughput sequencing assays (e.g., RNA-seq, ATAC-seq, ChIP-seq), which provide high-resolution count data that reflect how the phenotypes vary along the genome. Multiple methods have been proposed to help exploit these high-resolution measurements for differential expression analysis. However, they ignore the count nature of the data, instead using normal approximations that work well only for data with large sample sizes or high counts. Here we develop count-based methods to address this problem. We model the data for each sample using an inhomogeneous Poisson process with spatially structured underlying intensity function, and then, building on multi-scale models for the Poisson process, estimate and test for differences in the underlying intensity function across samples (or groups of samples). Using both simulation and real ATAC-seq data we show that our method outperforms previous normal-based methods, especially in situations with small sample sizes or low counts.
翻译:测算和测试不同条件下分子苯型(例如,基因表达、染色体可获取性、转录系数等)的差异,是了解基因调节的分子基础的一个重要部分。这些苯型通常使用高通量序列测算法(例如,RNA-seq、ATAC-seq、CHIP-seq)进行测量,这些测算和测试提供了高分辨率计数数据,反映了基因组中苯型的差异。提出了多种方法,帮助利用这些高分辨率测量法进行差异表达分析。但是,它们忽略了数据的计算性质,而没有使用仅对大样本大小或高数数据行之有效的正常近似值。我们在这里开发了解决这一问题的计数方法。我们用无异性皮森进程以空间结构化的强度功能对每个样本的数据进行模型建模,然后以多尺度模型为基础,估计和测试样品(或样品组)潜在强度功能的差异。它们忽略了数据的计算性质,而是使用通常的近似近似值或实际的ATAC值,特别是以先前的低位数计算方法,我们用以前的常规方法展示了以往的模型或低位数数据。