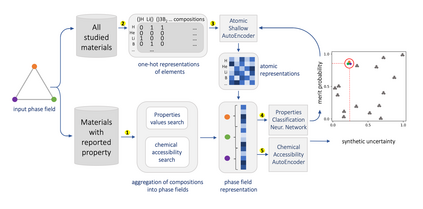
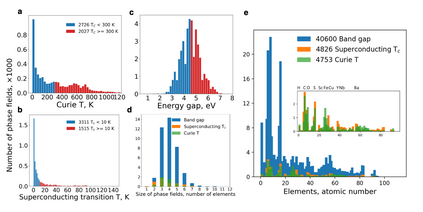
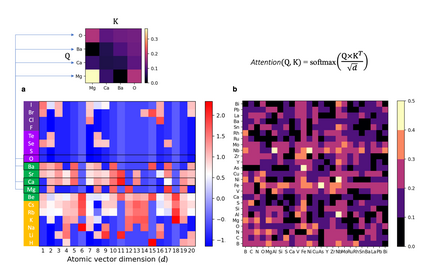
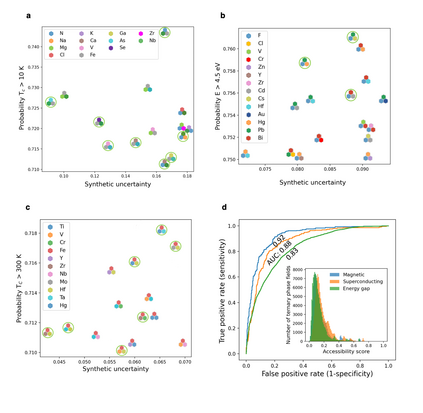
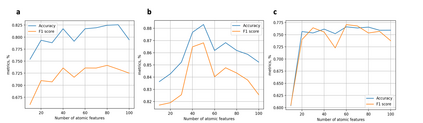
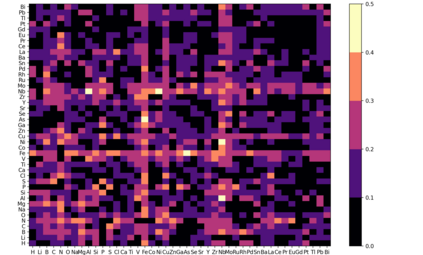
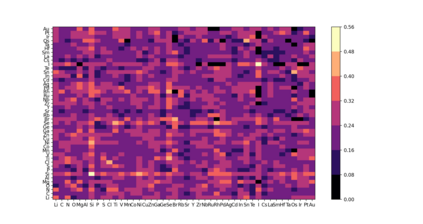
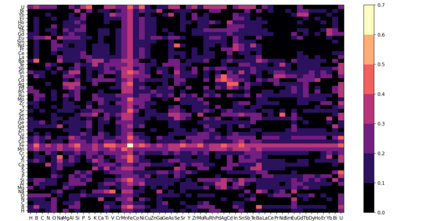
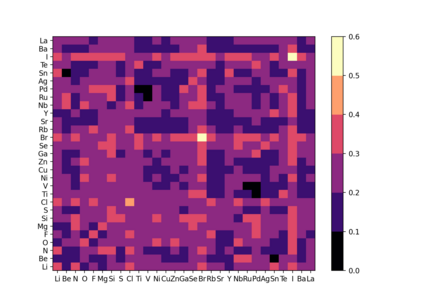
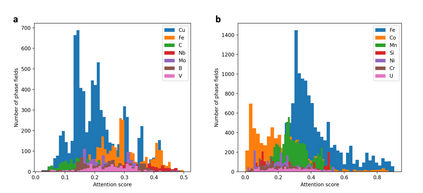
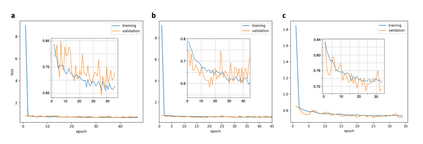
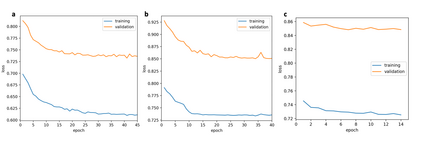
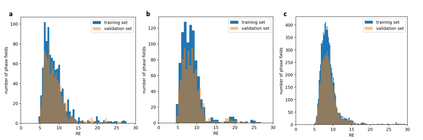
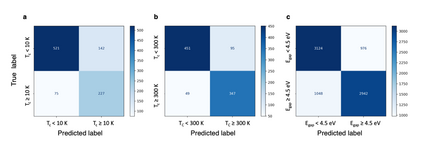
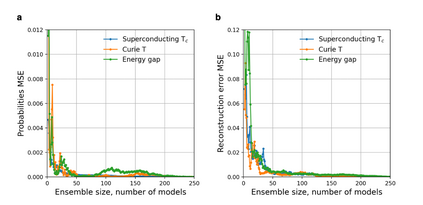
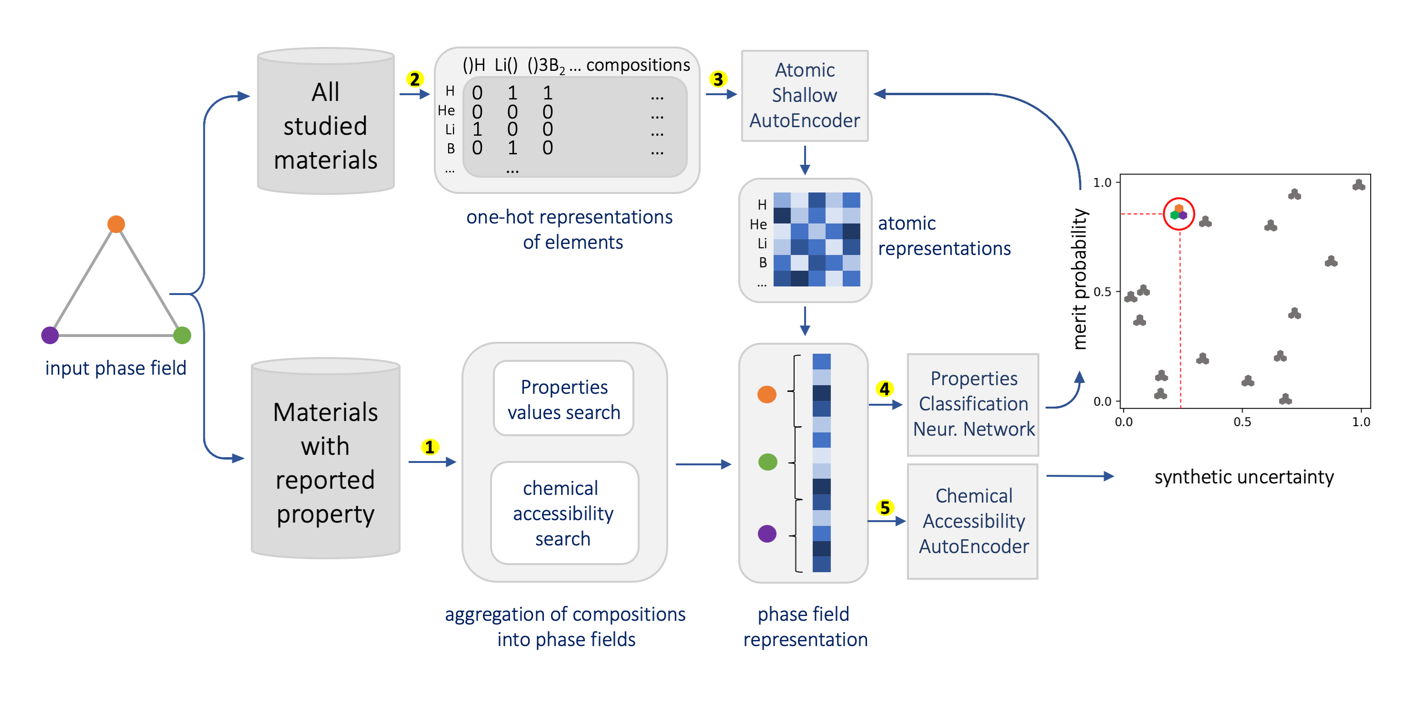
At the high level, the fundamental differences between materials originate from the unique nature of the constituent chemical elements. Before specific differences emerge according to the precise ratios of elements (composition) in a given crystal structure (phase), the material can be represented by its phase field defined simply as the set of the constituent chemical elements. Classification of the materials at the level of their phase fields can accelerate materials discovery by selecting the elemental combinations that are likely to produce desirable functional properties in synthetically accessible materials. Here, we demonstrate that classification of the materials phase field with respect to the maximum expected value of a target functional property can be combined with the ranking of the materials synthetic accessibility. This end-to-end machine learning approach (PhaseSelect) first derives the atomic characteristics from the compositional environments in all computationally and experimentally explored materials and then employs these characteristics to classify the phase field by their merit. PhaseSelect can quantify the materials potential at the level of the periodic table, which we demonstrate with significant accuracy for three avenues of materials applications: high-temperature superconducting, high-temperature magnetic and targetted energy band gap materials.
翻译:在高水平上,材料之间的根本差异源于组成化学元素的独特性质。在根据特定晶体结构(阶段)中元素(组合)的确切比率出现具体差异之前,材料可以以其阶段字段代表,仅定义为构成化学元素的一组。材料按其阶段字段的分类可以通过选择有可能在合成可获取材料中产生理想功能属性的元素组合加速材料的发现。在这里,我们证明,根据目标功能属性的最大预期价值对材料的分类可以与材料合成可及性的排序相结合。这种端对端机器学习方法(Scheste-theart)首先从所有计算和实验探索材料的构成环境中产生原子特性,然后利用这些特性根据它们的优点对阶段字段进行分类。阶段选择可以量化定期表格一级的材料潜力,我们用三种材料应用途径,即高温超导、高温磁力和定向能量波段材料,非常精确地证明了这些材料潜力。