分子性质预测的核心原则之一是相似性原则,但是分子对之间存在活性悬崖的情况(即分子结构相似但是活性却相差巨大的情况)。 2022年12月1日,埃因霍芬理工大学的研究团队在Journal of Chemical Information and Modeling上发表论文Exposing the Limitations of Molecular Machine Learning with Activity Cliffs。

在本文中,为了促进“以活性悬崖为中心”的模型的评估和开发,作者开发了Molecular ACE工具包。molecular ACE的目标是激励分子机器学习研究人员充分考虑长期存在的活性悬崖问题,在先导化合物优化过程中识别活性悬崖,推动准确预测细微结构变化对分子性质影响的模型的开发,提高先导化合物优化效率。这些改进将进一步推动深度学习在药物发现及其他领域的应用。 **1 摘要 **机器学习已成为药物发现和化学领域的重要工具。然而,活性悬崖问题 (activity cliffs,即分子对的结构高度相似,但在活性上表现出巨大差异的问题) 因对模型性能的影响而受到了有限的关注。本文的工作旨在填补当前在存在活性悬崖的情况下机器学习最佳实践方法的知识空白。作者对来自30个大分子靶标的精选生物活性数据进行了总共24种机器和深度学习方法的基准测试,以评估它们在活性悬崖化合物上的表现。尽管所有方法都在活性悬崖的存在下举步维艰,但基于分子描述符的机器学习方法优于更复杂的深度学习方法。
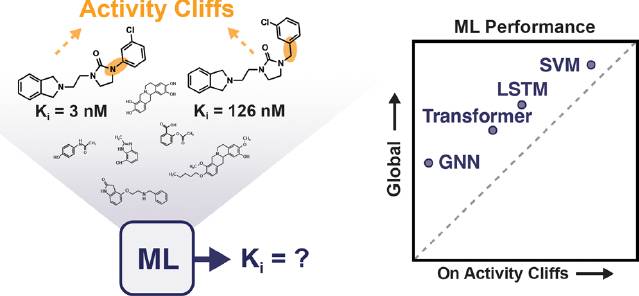
**本文的研究结果发现了模型在性能上的巨大差异,认为:(1) 需要在模型开发和评估过程中纳入专门的“以活性悬崖为中心”的指标;(2) 亟需开发新算法以更好地预测活性悬崖的属性。**为此,本研究的方法、指标和结果已封装到名为Molecular ACE (Activity Cliff Estimation) 的开放访问基准测试平台中,molecular ACE旨在引导社区解决活性悬崖造成的分子机器学习模型的紧迫、但被忽视的局限性。 molecular ACE可在GitHub上获得:https://github.com/molML/MoleculeACE). 2 数据与模型
2.1 数据集
为了确保模型性能的全面分析,作者收集并整理了来自ChEMBL v29数据库中的30个大分子靶标的数据。最终得到的数据集包含总共48,707个分子(其中35,632个是唯一的)。数据集包括多个靶标家族,如激酶、核受体、G蛋白偶联受体、转移酶和蛋白酶等,数据集也有大有小,具体情况如表1所示。 表1 数据集概览Inhibition, 抑制常数;Agonism, 半最大效应浓度EC50。总分子数,测试集分子数,活性悬崖总比例%cliff,活性悬崖测试比例%clifftest。

2.2 活性悬崖
对于每个大分子靶标,通过考虑分子对的结构相似性和效力差异来确定活性悬崖。作者用三种不同的方法量化了属于同一数据集的任何分子对之间的分子相似性: A. Substructure similarity子结构相似性。在扩展连接性指纹(extended connectivity fingerprints, ECFP)上计算了Tanimoto系数,以捕捉一对分子之间共享的径向原子中心子结构的存在。该方法通过考虑分子包含的所有子结构集来捕捉分子之间的“全局”差异(图1a)。 B. Scaffold similarity 骨架相似性,通过计算原子骨架上的ECFP并计算相应的Tanimoto相似度系数来确定。骨架相似性允许识别分子核心有微小差异或基于其骨架装饰不同的成对化合物(图1b)。 C. Similarity of SMILES strings 通过Levenshtein距离捕捉SMILES字符串的相似性。该指标检测字符插入、删除和移位(图1c)。
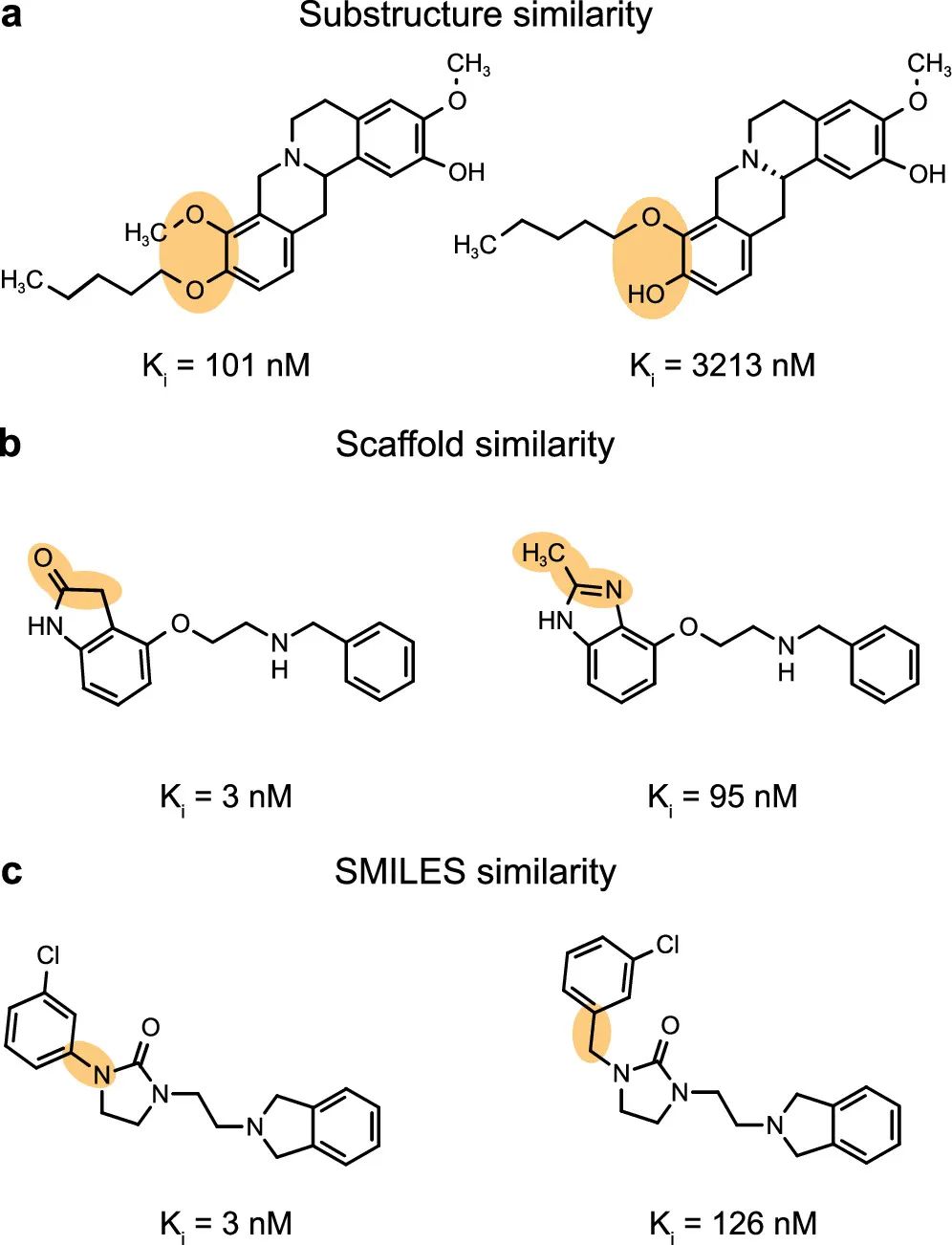
图1 展示的具有活性悬崖的实例(dopamine D3 receptor (多巴胺D3受体),D3R)。(a) 一般子结构相似性(ECFP上的Tanimoto系数)。(b) 骨架相似性,量化分子核心或支架装饰之间的相似性(骨架ECFP上的Tanimoto系数)。(c) SMILES相似性,检测字符串插入、删除和移位(规格化的Levenshtein距离)。 2.3 分子描述符的计算
作为计算模型的输入,作者利用RDkit软件基于SMILES计算了4种分子描述符,包括: A.扩展连接指纹(Extended connectivity fingerprints, ECFPs),1024 bits长度的0/1向量。B.MACCS keys,维度为166。C. 加权整体不变分子描述符(Weighted holistic invariant molecular, WHIM),114维。D.物理化学描述符,包括11种类药属性。 2.4 模型
作者总结了现有方法,建立了活性悬崖预测的基准模型,整理为了MoleculeACE工具包。工具包中的传统机器学习方法包括K近邻(K-nearest neighbor, KNN)、支持向量回归(Support vector regression, SVM)、梯度提升机(Gradient boosting machine, GBM)、随机森林(Random forest, RF),图神经网络方法包括图卷积网络(Graph convolutional network, GCN)、图注意力网络(Graph attention network, GAT)、消息传递网络(Message passing neural network, MPNN)、注意力分子指纹模型(Attentive fingerprint, AFP),前向深度学习模型包括卷积神经网络(Convolutional neural network, CNN)、长短时记忆神经网络(Long short-term memory (LSTM) networks)、Transformer模型。建立的所有模型如图2所示。
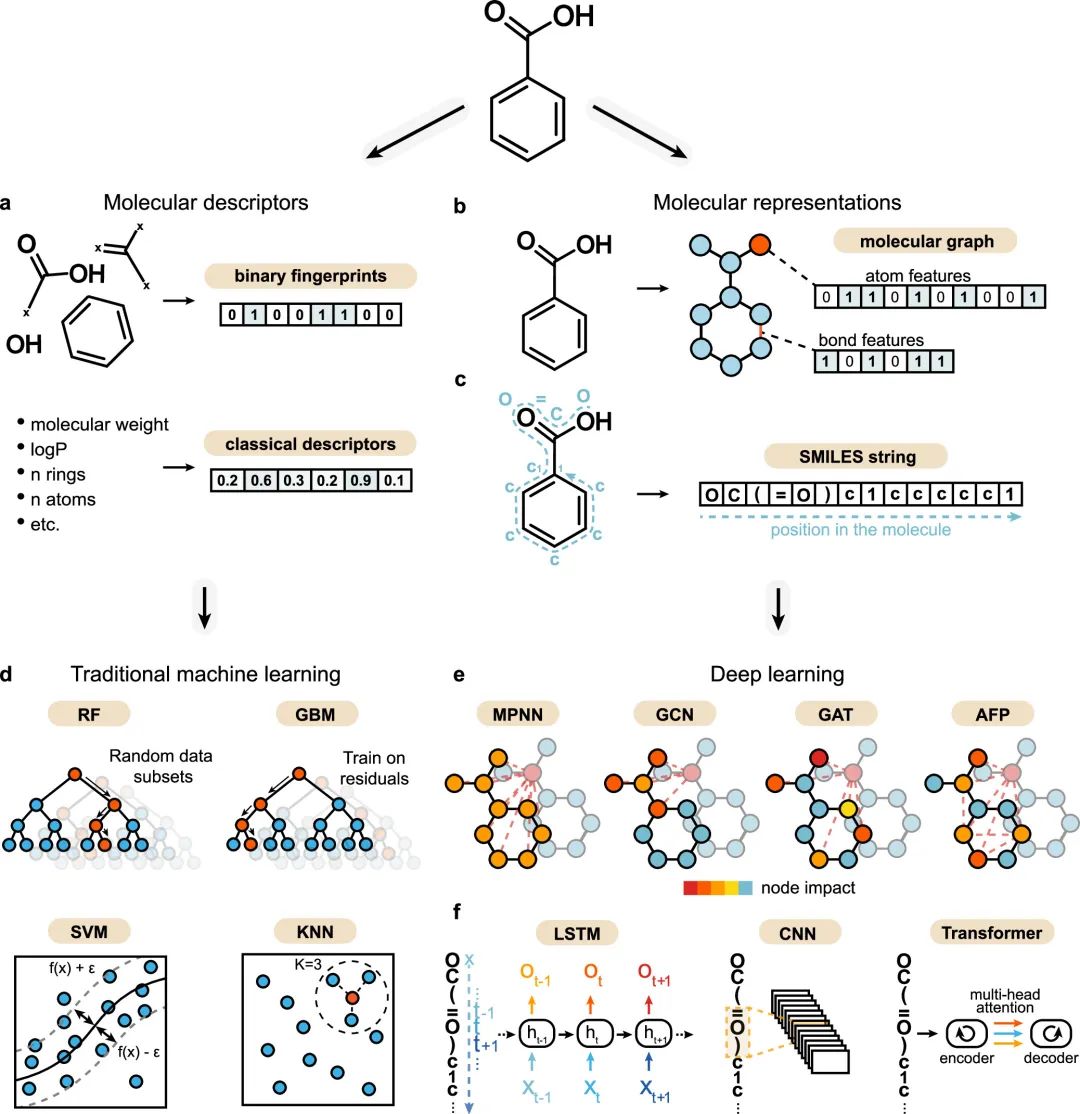
图2 机器学习方法汇总。(a) 分子描述符的简化表示,其捕获预定义的分子特征。本研究同时使用了二值分子指纹和传统的分子描述符。(b) 分子图,其中原子表示为节点(具有相应的节点特征),键表示为边(如果特征存在,则具有相应的边特征)。(c) SMILES字符串,具有二维信息(原子、键类型和分子拓扑结构)。(d) 选择了基于分子描述符训练的传统机器学习算法:随机森林(RF)、梯度增强机(GBM)、支持向量机回归(SVM)和K近邻(KNN)。(e) 深度学习方法。作者使用了四种可以从分子图中学习的图神经网络:消息传递神经网络(MPNN)、图卷积网络(GCN)、图注意力网络(GAT)和注意力分子指纹网络(attentive fingerprint, AFP)。节点颜色表示特征聚合期间其他节点对目标节点的影响(由虚线表示)。作者使用了三种可以从序列数据中学习的基于SMILES的方法:长短时记忆网络(LSTM)、一维卷积神经网络(CNN)和transformer模型。 2.5 模型结果
作者在论文中对基准方法进行了广泛的实验,对模型性能进行了比较和分析。图3展示了传统机器学习方法在预测任务上的表现。图4展示了深度学习方法在预测任务上的表现。图5展示了所有方法的整体模型性能和活性悬崖化合物性能的比较结果。
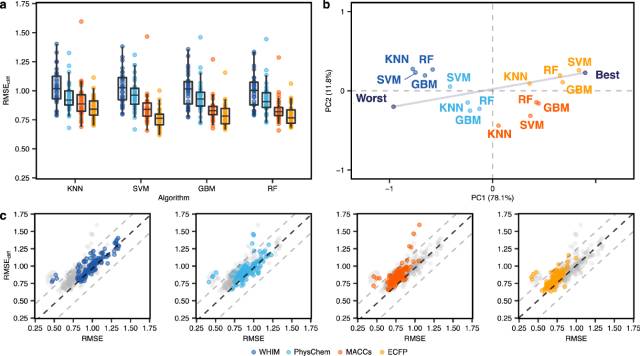
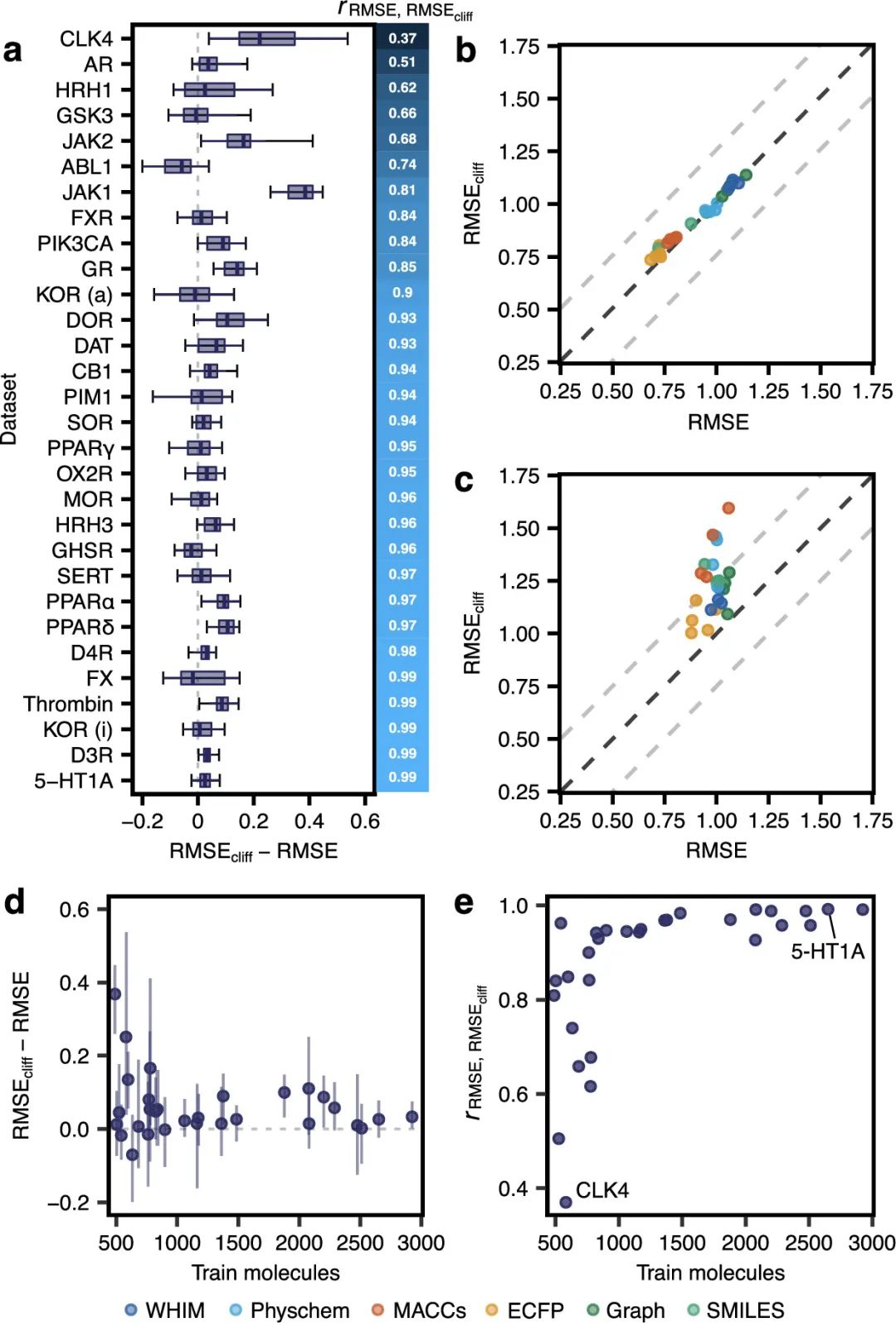
图3 传统机器学习方法的表现。(a) 使用不同机器学习算法和分子描述符(以颜色区分)在活性悬崖化合物上的RMSE结果。(b) 使用PCA (前两个主成分PC1和PC)对所有方法进行排序(已根据最好和最坏表现规格化)。每个点表示机器学习方法及其依赖的描述符的不同组合,并通过考虑所有数据集上的相应RMSEcliff来获得。“最差”和“最佳”分别表示在所有数据集中获得的最差和最佳性能。百分比表示每个主要成分所解释的差异。(c) 所有方法在活性悬崖化合物上的误差(RMSEcliff)和在所有化合物上的误差(RMSE)之间的比较。黑色虚线表示RMSE=RMSEcliff,而灰色虚线表示RMSEcliff和RMSE之间的差值为±0.5个对数单位。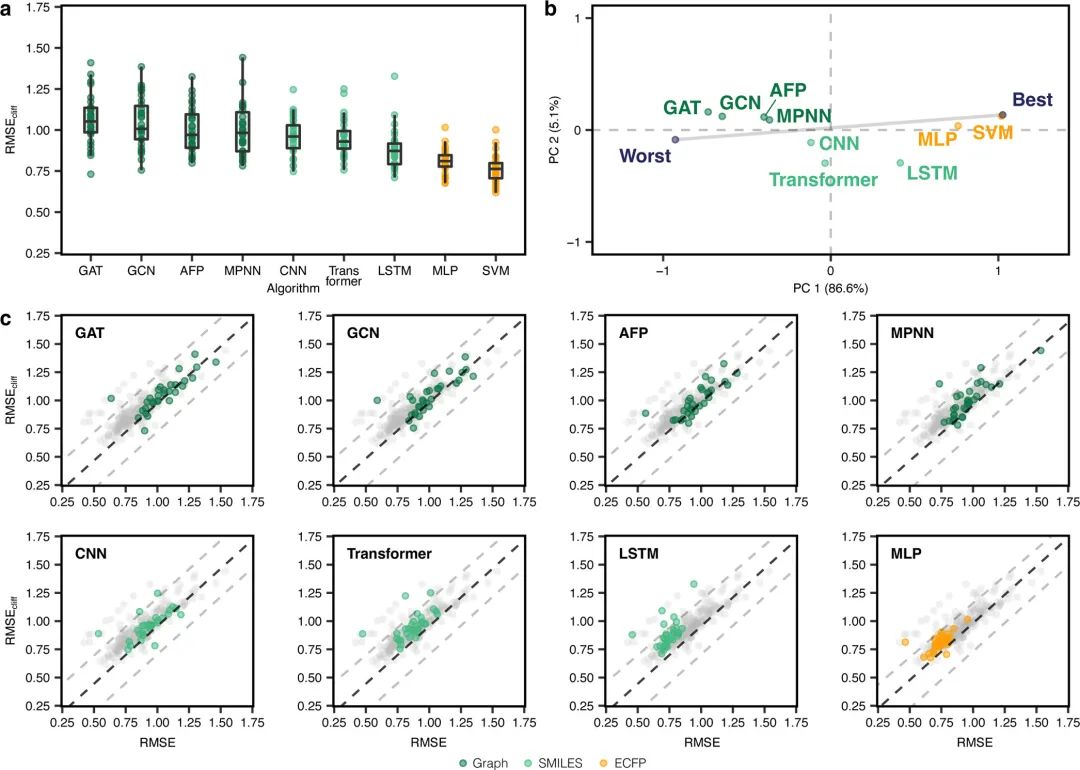
3 结论尽管机器学习越来越多地被用于早期药物发现,但活性悬崖这一话题在科学界的关注程度有限。正如结果所显示的,与整体表现相比,机器学习策略不仅难以应对活性悬崖,而且深度学习方法也特别受到这种问题的挑战。基于人工分子描述符的方法优于基于图或SMILES的深度学习,从它们的性能表现来看,目前没有任何机器学习策略在处理活性悬崖方面始终做到更好。本研究证实了先前的证据,表明深度学习方法不一定能与更简单的机器学习方法相抗衡。**尽管本文的分析不足以发现活性悬崖的性能差距的机制原因,但作者推测,当前的分子表示和相应的表示学习算法可能无法很好地捕获复杂的结构-活性信息。****作者设想如果开发有效的深度学习策略,该策略应该:(a)在少标签数据场景中更有效(例如,自监督学习),(b)****更适合捕获结构-活性“不连续性”,这是未来潜在应用的关键。**基于结构的深度学习方法(除了配体信息外,还考虑大分子靶标的结构)可能是填补由于活性悬崖导致的当前性能差距的关键。然而,迄今为止,在生物活性预测的机器学习中包含结构信息的益处尚未达成共识,这可能是由现有数据库中存在偏差导致的。在本研究设计框架中,模型在活性悬崖化合物上的性能出现了高度的数据集依赖性,特别是对于小数据场景中的深度学习方法。尽管总体预测误差通常接近活性悬崖上的表现,但当在同一数据集上比较不同策略时,活性悬崖上表现不佳的“孤岛”仍然存在。这些结果突出了评估机器学习模型在活性悬崖上的性能的重要性。 关于AI药物设计中的活性悬崖问题也可参见徐峻教授阐述:徐峻|人工智能辅助药物发现——从颠覆性思维到底层逻辑的重构
参考资料van Tilborg D, Alenicheva A, Grisoni F. Exposing the limitations of molecular machine learning with activity cliffs[J]. 2022. --------- End ---------