
导语
物种进化和分子进化是进化生物学研究的两个重要主题,它们之间是否存在着某种联系?近日,日本理化学研究所研究科学家、集智科学家唐乾元等人,利用人工智能系统AlphaFold预测的蛋白质结构,通过统计物理分析,揭示出随着生物体复杂度的提高,生物体的基本组成单元——蛋白质分子在进化中所表现出的统计趋势。相关成果发表在分子生物学和进化生物学领域的知名期刊Molecular Biology and Evolution。
研究领域:生物进化,生物复杂度
傅渥成 | 作者****
邓一雪** **| 编辑

论文题目:The Statistical Trends of Protein Evolution: A Lesson from AlphaFold Database论文链接:https://academic.oup.com/mbe/advance-article/doi/10.1093/molbev/msac197/6701686
1. 物种进化与分子进化
尽管并不存在着某种固定的“进化方向”,但从宏观尺度来看,的确可以看到生命的复杂性在漫长的进化过程中不断增加,例如从原核生物到真核生物,从单细胞生物到多细胞生物等等。在微观尺度下,与生物体的复杂化并行发生着另一种进化过程,那就是分子进化,即作为生物体基本构件的蛋白质分子也在不断进化。进化的这种宏观(生物体)与微观(蛋白质分子)视角之间是否存在某种联系?直观上来看,某种特定的蛋白质分子的进化不一定遵循物种进化的路径,然而,如果把视角扩展到大量蛋白质的集合、甚至是一个生物体内的全部蛋白质,或许能从中挖掘出某些集体特征,反映出与生物体复杂性相一致的统计趋势。
上述这种宏观与微观之间的联系与经典的统计物理问题类似:从微观出发,观察气体分子的运动,会发现其运动杂乱无章,看似毫无秩序;若是切换到宏观视角,将整个系统用少数几个热力学量(如压强、温度等)来描述,则能发现系统某种“集体性”的演化趋势。如果能从大量的微观个体的演化(即蛋白质的进化)中提取出与系统宏观演化方向(即物种演化)相一致的趋势,就能对生命的起源和进化问题有全新的认识。不过在很长的一段时间里,由于已知的蛋白质结构仍然非常有限,难以真正讨论物种体内蛋白质整体进化趋势。幸运的是,最近人工智能的发展为研究者们提供了全新的强大的工具,让上述研究思路能够真正得以实现。
近日,日本理化学研究所(RIKEN)唐乾元(aka 傅渥成)博士和任卫同博士、与南京大学王骏教授、丹麦哥本哈根大学金子邦彦(Kunihiko Kaneko)教授合作,基于人工智能系统AlphaFold 预测的蛋白质结构,通过统计物理分析,揭示出随着生物体复杂度的提高,生物体的基本组成单元——蛋白质分子在进化中所表现出的统计趋势。这一研究是首次对40多种生物体内蛋白质组内的全部蛋白质结构进行统计分析,该工作发表在分子生物学和进化生物学领域的知名期刊 Molecular Biology and Evolution(2021 IF: 8.8,中科院1区Top)。
**
**
2. AlphaFold数据库
2021 年,Science 和 Nature 杂志不约而同地将“年度十大科学突破”颁给了由 Google DeepMind 开发的蛋白质结构预测系统 AlphaFold 2。AlphaFold作为DeepMind开发的人工智能系统,能够利用共进化(coevolution)信息提供高准确度的蛋白质结构预测,并且在此前的蛋白质结构预测竞赛中中赢得了前所未有的压倒性成功 [1, 2]。从2021年开始,AlphaFold发布了自己的蛋白质结构数据库(AlphaFold Protein Structure Database,以下简称AlphaFold DB),其中包含了从细菌、古细菌、单细胞和多细胞真核生物到人类等在内的许多物种的完整蛋白质组,这个数据库还在不断扩大,在2022年7月底的更新中,AlphaFold DB已经扩充到包含约2亿个预测的蛋白质结构 [3, 4]。AlphaFold DB不仅能帮助科学家们解决医学和生命科学中的关键问题,而且在进化研究中显示出了新的可能性。
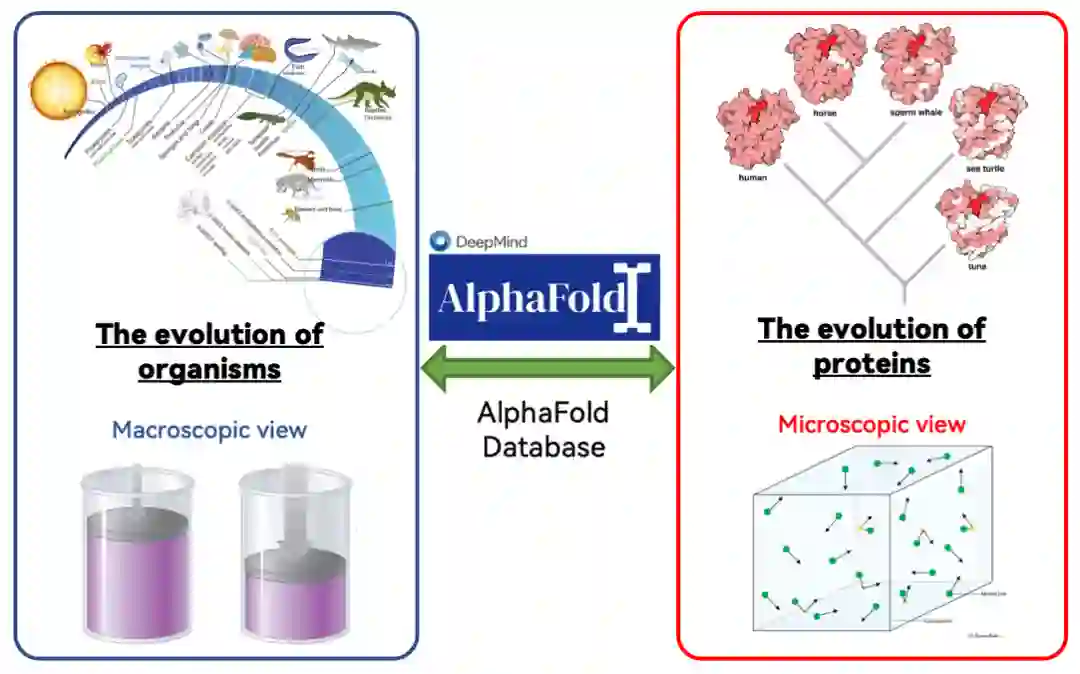
图1. 研究思路示意图。利用AlphaFold 蛋白质结构数据库,建立起物种进化与分子进化之间的联系。
与传统的分子进化研究不同,唐乾元博士及其合作者利用 AlphaFold DB 发展了一套基于物种全蛋白质组蛋白结构的进化分析方法,对不同生物体内的全部蛋白质进行统计性的研究,而不是只关注特定的蛋白质家族。研究者们从序列、结构、氨基酸残基的拓扑、蛋白质平衡态的动力学等角度出发,揭示了随着物种朝着更为复杂化的方向进化、物种体内的蛋白质呈现出的整体进化趋势。下面,本文将主要从物理学图像的角度出发,简要介绍这些进化中的统计趋势。如果你对关于生命复杂性的延伸讨论更感兴趣,可以直接跳到本文的第6小节。如果你对于相关的研究细节与分析方法感兴趣,除了阅读第3~5节的讨论以外,也可以点击文章开头的“论文链接”,直接阅读论文。
**
**
3. 结构柔性
研究者们首先对不同生物体内、链长相近的蛋白质分子的结构进行了对比分析。尽管选取的这些蛋白质链长接近,但在不同生物体内,这些蛋白质分子的回转半径(radius of gyration)分布却非常不同(如图2A所示)。例如,在大肠杆菌(E. coli)体内链长约为250个氨基酸的蛋白质,平均的回转半径大约为20 Å,而在人类体内相近链长的蛋白质的平均回转半径却接近30 Å,两个复杂度差异巨大的物种体内的蛋白质半径分布也有显著的统计差异。由于是在对相近链长的蛋白质进行比较,这时,更大的回转半径主要是跟蛋白质结构中结构涨落较大的柔性片段相关,因此,这一结果还表明人体内的蛋白质比大肠杆菌体内蛋白质有更高的柔性。
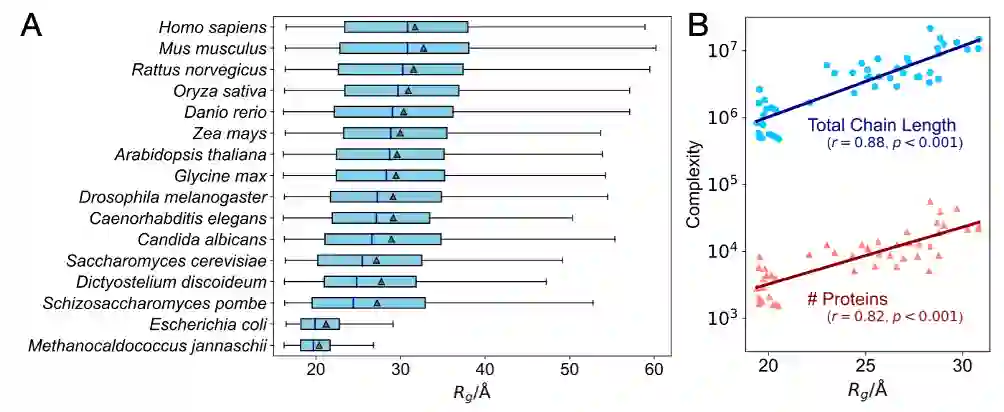
图2. 随着物种复杂度的增加,其体内一定链长的蛋白质的回转半径(反映结构的柔性)会相应增加。
对不同物种体内的链长相近的蛋白质结构进行统计,会发现一个粗略的相关关系:随着物种复杂度的提高,该物种体内的蛋白质的回转半径相应地会出现增大的趋势。这里涉及到了“复杂度”的概念,尽管复杂性的数学定义仍有争议,但大家对于生物体本身的复杂性仍然会有许多直观的理解(例如真核生物比原核生物更复杂)。在实际操作中,生物学家们往往会对生物体的复杂性引入不同的衡量标准,例如生物体内的各种细胞类型的总数、基因组大小、蛋白质组大小等等。这些定义分别侧重于生物复杂度的不同层面,这些不同的度量之间往往也是相互关联的 [5]。在本研究中,研究者们基于蛋白质组的数据,引入了:(1)一个生物体内所有的蛋白质种类数,以及(2)各种不同蛋白质的总链长作为生物复杂性的度量。如图2B所示,这两种生物复杂度的度量都与一定链长的蛋白质的回转半径成正比,证明随着物种复杂度的提高,其体内的蛋白质表现出更高的柔性。
在论文中,研究者不仅讨论了其它链长的情况,还对AlphaFold预测的结构精度进行了进一步的筛选,而且还对蛋白质的二级结构(常见的二级结构包含α螺旋、β折叠、无规卷曲等)进行了分析,进一步验证了“生物体复杂度与其体内蛋白质的平均柔性成正相关关系”的结论。
**
**
4. 序列和拓扑
要更深入地分析蛋白质的结构,除了对蛋白质的二级结构、三级结构进行分析以外,也可以将蛋白质视为氨基酸残基在空间中相互靠近接触而形成的网络,用网络拓扑分析的方法来分析蛋白质的性质。在残基接触网络(residue contact network)中,每个节点所代表一个氨基酸残基,残基空间距离小于一定的截断长度的,则被视作存在连边。在论文中,研究者们对于这个网络的许多拓扑性质进行了分析,其中与蛋白质的物理性质最为相关的度量是网络的同配性(assortativity)。
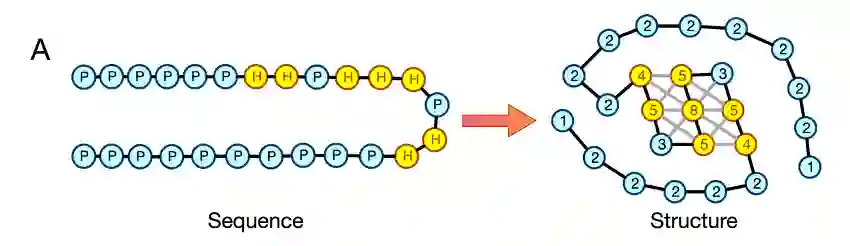
在一个复杂网络上,如果那些度数(连边数)较大的节点倾向于跟度数同样较大的节点相连接,那么这样的网络就是同配的。举个例子,假如在一个社交网络上,各种大V用户抱团取暖,相互关注,而各种普通用户只能跟自己同样是普通用户的三五好友相互关注,这样的网络就是同配的。反之,如果度数较大的节点倾向于跟度数较小的节点连接,例如在一个社交网络上,一个大V可能吸引到许多低关注数的普通用户关注,这样的网络就不是同配的。蛋白质的残基接触网络是高度同配的,这是因为构成蛋白质的氨基酸残基可以被分为“亲水”和“疏水”两类,疏水氨基酸残基往往被包埋在蛋白质的内部,形成紧密的堆积,而亲水氨基酸残基则暴露在蛋白质的表面,甚至可能形成高度柔性的卷曲(如图3右所示)。
对AlphaFold预测的蛋白质结构进行统计,研究者们发现,生物体复杂度与其体内蛋白质的残基接触网络的平均同配性成正相关关系。这一结果与上一节讨论的统计趋势也是自洽的,因为同配性的残基接触网络让亲水和疏水氨基酸残基在空间上产生了分隔,导致“贫者越贫”,蛋白质结构中出现了更多高度柔性的无序片段,蛋白质的回转半径也因此增加。
在观察到蛋白质残基接触网络的拓扑性质在进化中出现的统计趋势之后,研究者们又进一步对蛋白质的序列进行了统计。如图3所示,蛋白质的三维结构由其序列所决定,那么,到底是序列上的什么特征导致了蛋白质残基接触网络的同配性呢?研究者们发现,亲水和疏水氨基酸残基在三维空间中的分隔,与其在序列上的分隔是相关的。换言之,如果一个蛋白质的序列出现了大段的连续亲水或者连续疏水氨基酸,这样的序列将更容易形成高度同配的残基接触网络。研究者们观察到,随着物种复杂度的增加,序列中亲疏水氨基酸的分隔的确有逐步提升的趋势。
需要特别强调的是,上述的序列分析完全不依赖AlphaFold的结构预测,而从序列结构中所揭示的统计趋势又可以在很大程度上支持结构和拓扑分析的结果。这些结果表明,论文所讨论的“蛋白质进化的统计趋势”并非是由结构预测方法所带来的系统偏差,而是的确反映了某种自然趋势。
5. 功能专一性
蛋白质的生物功能是由其结构所决定的。上文所讨论的序列、拓扑和结构变化毫无疑问会影响蛋白质的生物功能。那么,蛋白质的生物功能会随着物种复杂度的提升,产生怎样的统计趋势呢?
为了研究这一问题,研究者对残基接触网络的拉普拉斯矩阵(graph Laplcaian)进行了谱分析,这种分析方法其实就是基于弹性网络模型的简正模分析。我们曾经在《为什么蛋白质兼具可塑性与稳定性?从进化视角揭示生命复杂系统的内在平衡》这篇文章中介绍过相关的分析方法。简而言之,蛋白质的运动可以被简化为其在天然态(能量最低结构)附近的振动,而这种振动可以由一系列的“振动模式”的叠加来描述。拉普拉斯矩阵的特征值(eigenvalue)正比于蛋白质分子振动模式的频率的平方,而与这些特征值相对应的特征向量(eigenvector)则描述了相应振动模式的基本形态(各个氨基酸残基会朝着怎样的方向、以怎样的相对振幅运动)。在拉普拉斯矩阵的特征值谱中,越小的特征值反映的是氨基酸残基更为低频、大振幅的集体运动,而越大的特征值反映的则是高频、小振幅的局域运动。或者反过来,如果关注拉普拉斯矩阵特征值的倒数,即拉普拉斯矩阵逆矩阵中较大的那些特征值,这实际上等效于进行主成分分析(principal component analysis, PCA)。这一方法常常被用于分析蛋白质平衡态运动中的“主成分”。
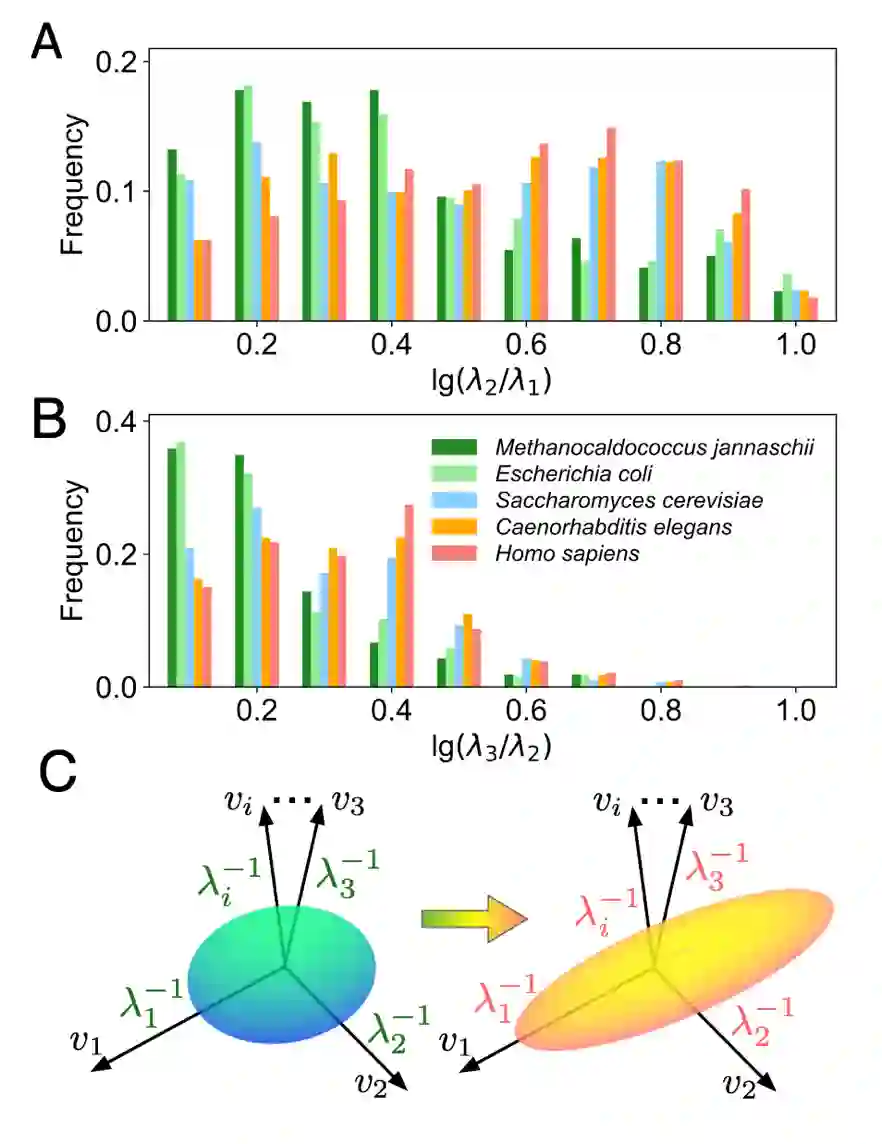
图4. 随着物种复杂度的提高,生物体内蛋白质动力学中相应主成分的比例会发生变化,蛋白质结构变化的空间会出现“降维”的趋势。
对不同物种体内的蛋白质进行振动谱分析,研究者们发现,随着物种复杂度的提升,蛋白质平衡态运动中的主成分比例会发生相应的变化。例如同样链长的蛋白质,在大肠杆菌中,它运动的第1主成分跟第2主成分之间的相对大小较为接近,而在人体中,它的第1主成分和第2主成分之间会有较大的区别(如图4A所示)。进一步的分析发现,随着物种由简单到复杂,其体内的蛋白质分子的动力学会出现“降维”的趋势,即运动的第1主成分会与第2主成分之间拉开越来越大的差距,第2主成分会跟第3主成分之间拉开越来越大的差距(图4B),以此类推。这种动力学中的“降维”趋势让蛋白质特定的功能运动模式变得更加突出(如图4C所示)。在复杂度更高的生物体内,有更多蛋白质倾向于沿着特定的主成分方向发生功能运动,这种特定的主成分方向往往与特定的功能有关。简而言之,随着物种从简单到复杂,构成生物体的蛋白质呈现出从“通用”到“专用”的统计趋势,高复杂度的生物体内更可能出现高度功能专业化的蛋白质。
**
**
6. 延伸讨论
蛋白质的“功能专业化”和生物体的复杂性之间的统计相关性与此前大量生物化学实验观察结果一致。许多研究表明,祖先酶往往具有更高的混杂性(promiscuity),即它们不仅可以催化主反应,还具有催化副反应的能力,利用祖先序列重建的方法,有助于设计具有高热稳定性和高混杂性的酶。
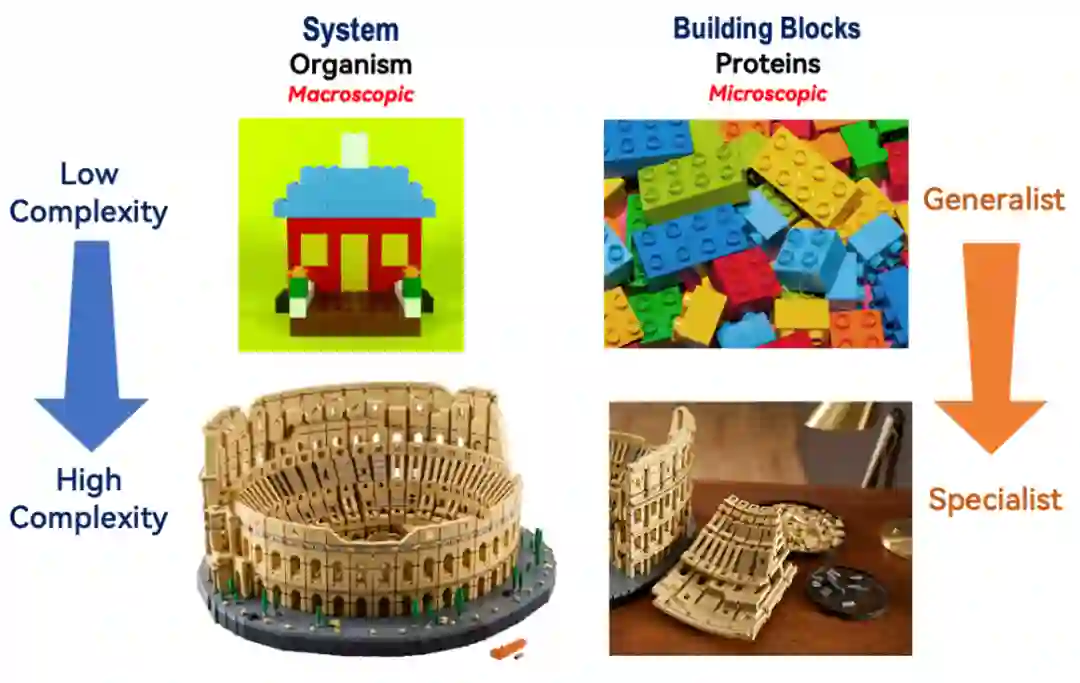
图5. 随着物种的复杂度提高,其体内的蛋白质分子有某种从“通用”向“专一”方向进化的统计趋势。这种现象并不是生物体系的某种特例,而是复杂系统中具有普适性的某种设计原则。
值得一提的是,祖先酶的热稳定性和高混杂性与祖先物种的低复杂性是相匹配的。复杂性低的生物体的基因组相对较小,其体内所包含的酶的种类也较少。尽管基因组规模小,但高混杂性的酶可以帮助这些生物体实现各种生命活动。相反,较大的基因组可以编码更多的蛋白质,能够发挥高度专业化的功能,应对更复杂和多样化的细胞环境。蛋白质的专业化和多样化使它们能够在更复杂和多样化的细胞环境中发挥作用。因此,复杂的生物体可以更有效地发挥其生物功能,获得适应复杂和多样化的外部环境的可塑性。
生物体的复杂性和组成蛋白质的功能专业化之间的兼容性不是生物体系的某种特例,而是复杂系统中具有普适性的某种设计原则(design principle)。复杂系统的整体和部分之间是相协调的。当一个系统变得更加复杂时,其组成部分或元素应该改变其属性(例如变得更加可塑或模块化)。一个直观的例子如图5所示,用乐高玩具搭建不同复杂度的建筑,如果只需要搭建一个简单的“建筑”,只需要用很少的几种元件就能完成,我们甚至可以交换某些元件;然而搭建一个复杂的结构时,不仅元件的总数大大增加,而且各种元件的通用性也在不断降低(即在一个乐高拼图中,两个形状接近的元件就不再能交换使用了)。在经济生活中,也有很多类似的现象。一家工厂,如果资金不足,但临时又需要生产某种新产品,往往会对原来的生产线进行改装——这也是某种“非专一性”的体现;如果这家工厂有了充足的资金,它可以投资购入一条全新的生产线,专门生产新产品,此时,虽然成本有所提高,但更为专一的生产线生产效率往往也会更高。
当然,需要强调的是,本文所讨论的各种“趋势”都是统计性的,它所反映的是一种整体趋势,而不是某种放之四海而皆准的法则(例如,也可以找到一些酶,它们具有更高的柔性,同时也有较高的混杂性)。总之,具体到每一种蛋白质分子,在定向进化和设计的过程中,都需要具体问题具体分析。
**
**
7. 总结与展望
这篇论文利用各种统计物理方法对AlphaFoldDB进行了全面的分析,展示出蛋白质进化中的统计趋势,即:随着生物体向更高的复杂性进化,其体内的蛋白质在统计意义上倾向于向更高的灵活性、更高的结构多样性进化,分子本身的功能专一性也在不断增强。除了在这篇文章中所讨论的一些内容以外,在原论文中,还对残基接触网络的模块度、残基堆积的分形维度、谱分布的Zipf指数等进行了分析。未来,基于人工智能预测的蛋白质结构的蛋白质组分析,与其他类型的生物信息(如蛋白质与蛋白质的相互作用网络、蛋白质的表达水平、进化速度等)相整合,必将为我们提供对细胞和生物体的行为和进化提供全新的见解。
延伸阅读
蛋白质的动力学和进化之间的对应关系:两个不同时间尺度下的相同故事 * 为什么蛋白质兼具可塑性与稳定性?从进化视角揭示生命复杂系统的内在平衡 * 博士招生 | 香港浸会大学物理系理论与计算研究团队:统计物理,机器学习,计算生物学,脑科学方向
参考文献1. Senior AW, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson AWR, Bridgland A, et al. 2020. Improved protein structure prediction using potentials from deep learning. Nature 577:706–710.2. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589.3. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A, et al. 2022. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 50:D439–D444.4. AlphaFold reveals the structure of the protein universe: https://www.deepmind.com/blog/alphafold-reveals-the-structure-of-the-protein-universe5. Niklas KJ, Cobb ED, Dunker AK. 2014. The number of cell types, information content, and the evolution of complex multicellularity. Acta Societatis Botanicorum Poloniae 83:337–347. 点击“阅读原文”,跳转论文原文************************
