作者 | 于洲
多重组学技术在生物医学中的应用能够揭示患者水平的疾病特征和对治疗的个体化反应。然而,传统的数据解释方法不足以充分利用多模态数据,多模态数据的规模和异构性质使得数据的整合和挖掘面临困难。
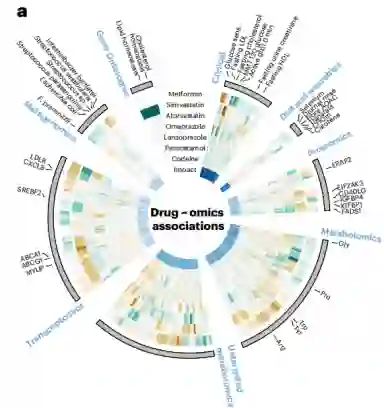
对此,哥本哈根大学Søren Brunak和Simon Rasmussen团队以及IMI DIRECT联盟开发了一个基于深度学习的框架——多组学变分自编码器(MOVE),该模型用于整合这些数据。作者团队将其应用于789名新诊断的具有深度多组学表型的Ⅱ型糖尿病患者数据,探究药物组学相关性。作者在多模态数据集中确定了20种最常见的药物组学相关性,发现了二甲双胍和肠道微生物群之间的新联系,以及辛伐他汀和阿托伐他汀两种他汀类药物的分子反应,最终得出药物效应分布在多组学模式中的结论。

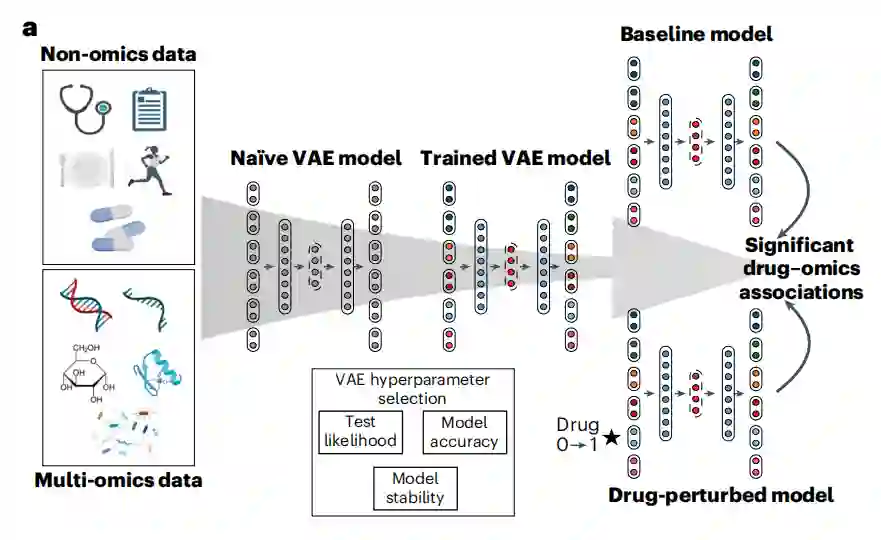
在之前的研究中,作者在变分自编码器(VAE)的基础上开发了一个深度学习框架,用于大量非结构化宏基因组数据的集成和分箱,发现VAE可以在没有任何先验知识或统计模型的情况下学习整合两个数据集。该框架利用了VAE的解码器作为一个生成模型,最终训练的解码器将能够从学习的潜在分布中生成新的数据示例。对于作者的多模态数据,作者假设VAE的生成能力将允许作者识别患者暴露和组学特征之间的关联。MOVE方法能够将多组学数据与临床和分类数据整合在一起,并且能够抵抗数据中的系统性偏差以及大量缺失数据。作者比较了药物的多组学特征,发现不同的药物具有独特的临床和分子特征。该MOVE是免费提供的,易于扩展,可以集成任意数量的分类和连续数据集,并能够识别多组学关联的特征。
实验结果
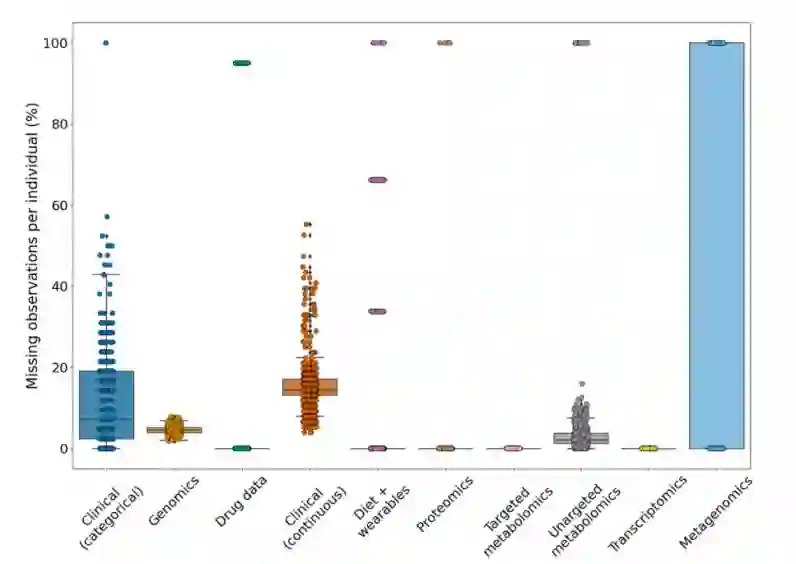
设计VAE 作者使用了789个新诊断的T2D个体的数据集,具有广泛的多组学特征。在组学数据集中,每个个体总共包含8807个变量,中位缺失量小于5%,宏基因组数据除外,其中三分之二的个体(532)没有任何数据。因此,这些个体在多组学数据中的缺失量高达24.7%。对于临床数据缺失较高,连续和分类临床数据的个体中位数分别为14%和7%。
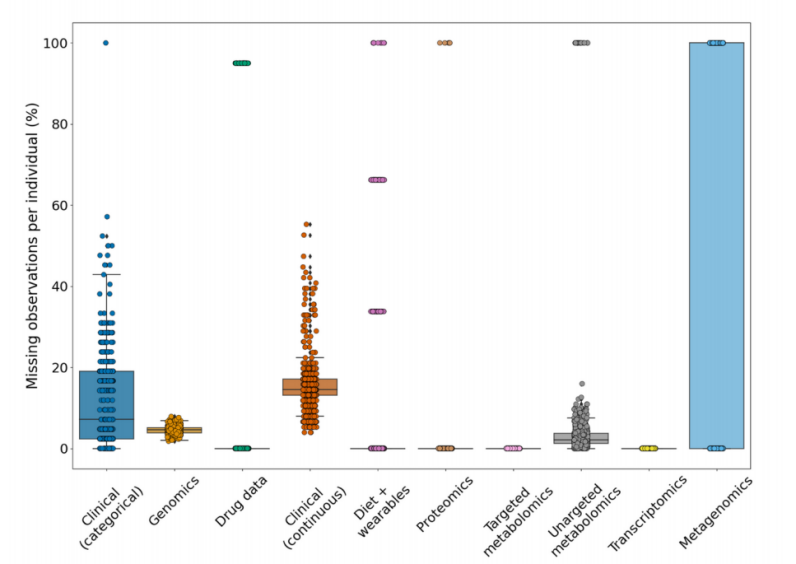
图:数据集之间缺少数据
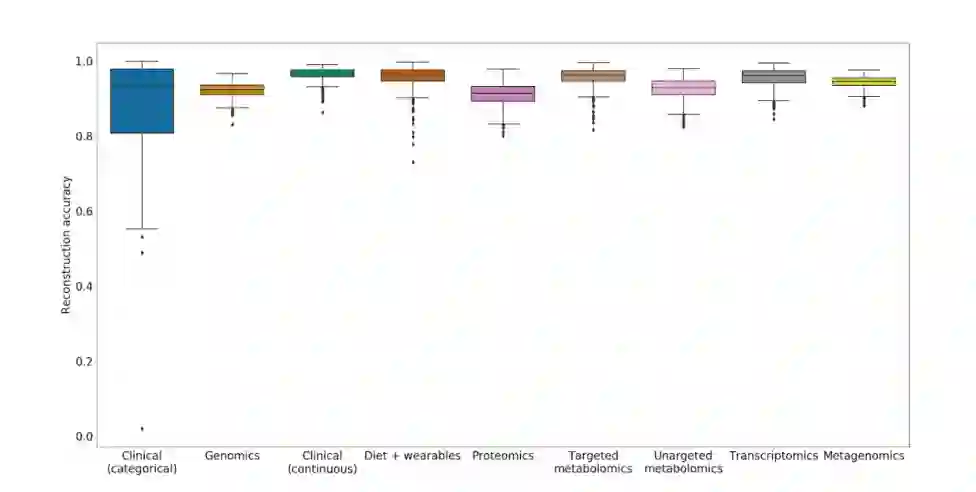
对此,作者设计的MOVE框架在输入数据类型的数量方面是灵活的,并且能够处理连续和分类特征。为了确定最优的超参数,既能捕捉数据的结构,又不会失去对未见过的个体进行泛化的能力,作者将数据集分为训练集和测试集。然后,作者测量了模型重构输入的能力,以及将模型多次改装到数据时的稳定性。重建精度的中位数在0.95-1之间,当重新训练5次时,最终模型高度稳定,潜在空间余弦相似度的平均变化为0.037。这证明VAE模型能够在个体之间以较高的精度重建数据。
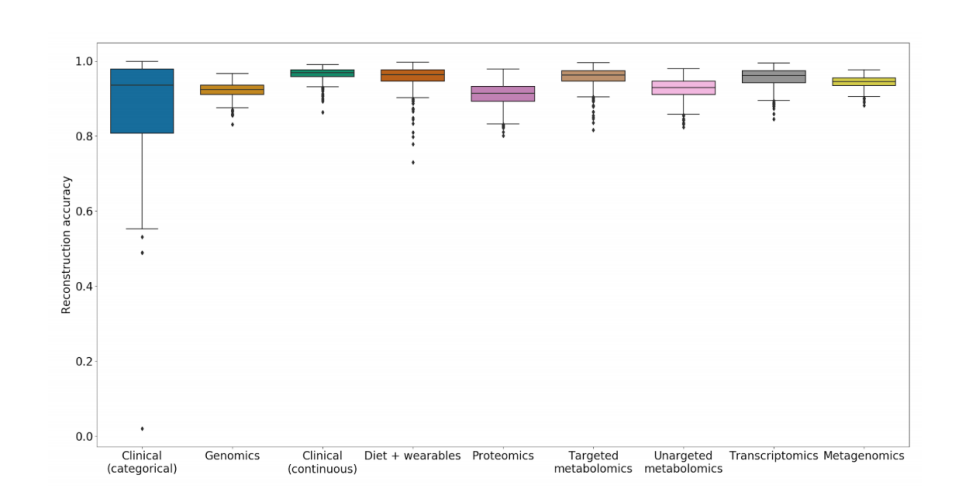
图:所选VAE模型的重构精度
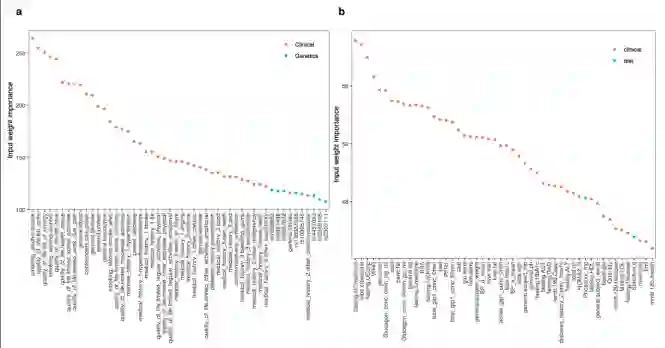
潜伏空间中重要的临床特征 为了说明该模型如何很好地捕获临床数据的结构,作者分析了连接到编码器输入变量的神经网络权重。在这里,作者发现大多数临床和饮食变量都是最重要的50个变量之一。
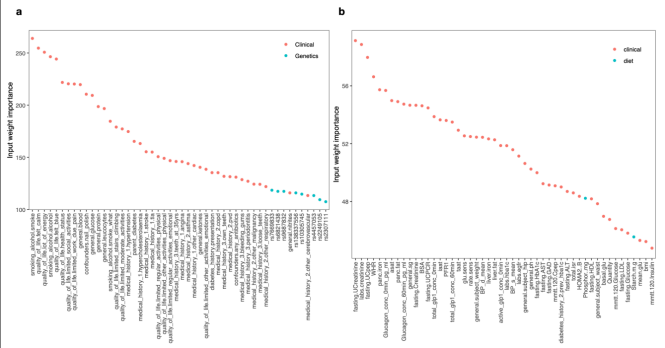
图:MOVE第一层与潜在特征重要性
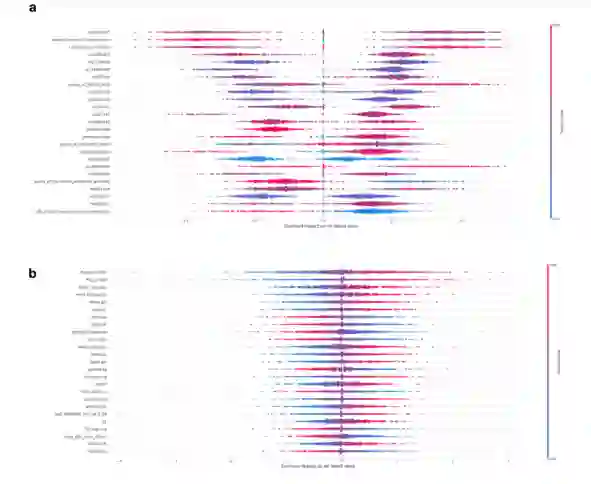
当作者使用Shapley相加解释(SHAP)分析研究连续特征如何影响个体在潜在空间中的定位时也是如此,而对于离散特征,作者发现t2d相关的遗传变异以及临床相关的特征都很重要。
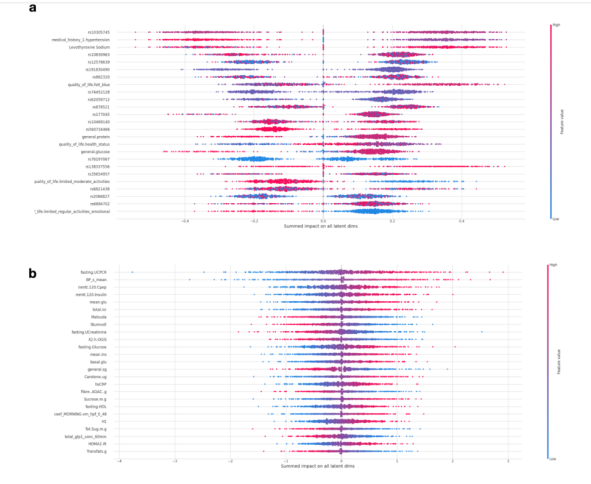
图:MOVE潜在空间的SHAP分析
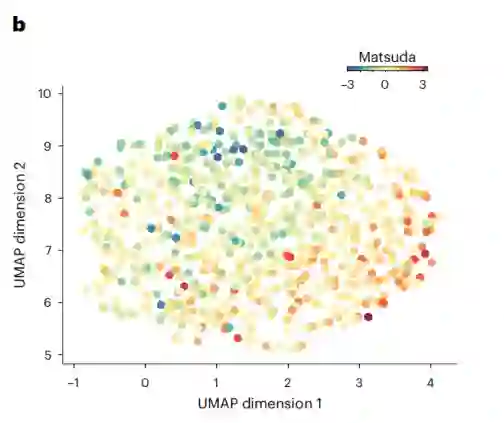
作者也研究了个体如何通过Matsuda指数量化的胰岛素敏感性等特征进行区分。
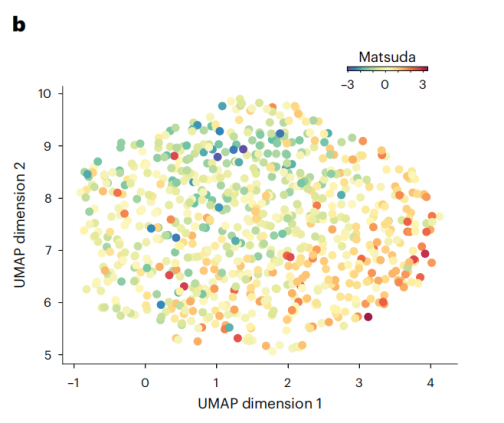
图:患者的潜在UMAP表现为潜在表现(根据个体的z-scale Matsuda指数进行着色)
作者还使用KNN分类器来研究混杂因素性别和招募中心对潜在表征的整体结构的影响。这些对性别和营救中心分别实现了0.58和0.25的精度,应与分别为0.50和0.17的偶然精度进行比较。这证明了VAE集成异构数据的能力,但也证明了大量的混杂因素可以影响模型潜在的表示。
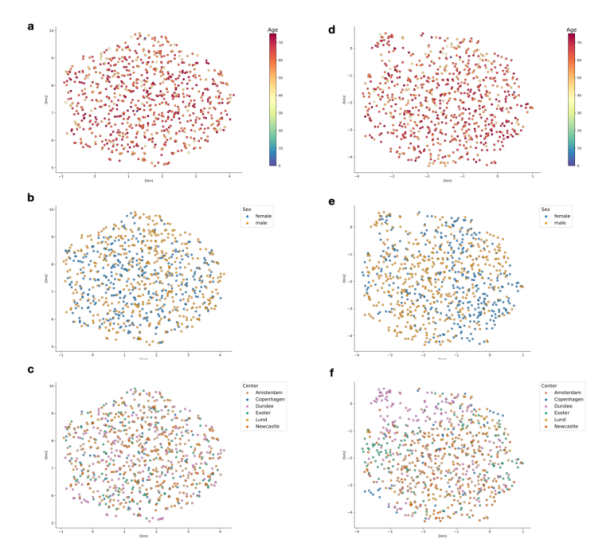
图:残差数据与非参数差数据在MOVE下的潜在UMAP能抵抗数据混淆
提取药物临床与多组学的关联 作者调查了该模型是否学习了临床、药物和多组学数据之间的关联。作者开发了一种基于每次扰动输入特征的方法。然后,作者评估了与将原始数据通过模型时相比,观察每个特征重建的变化是否有显著差异。
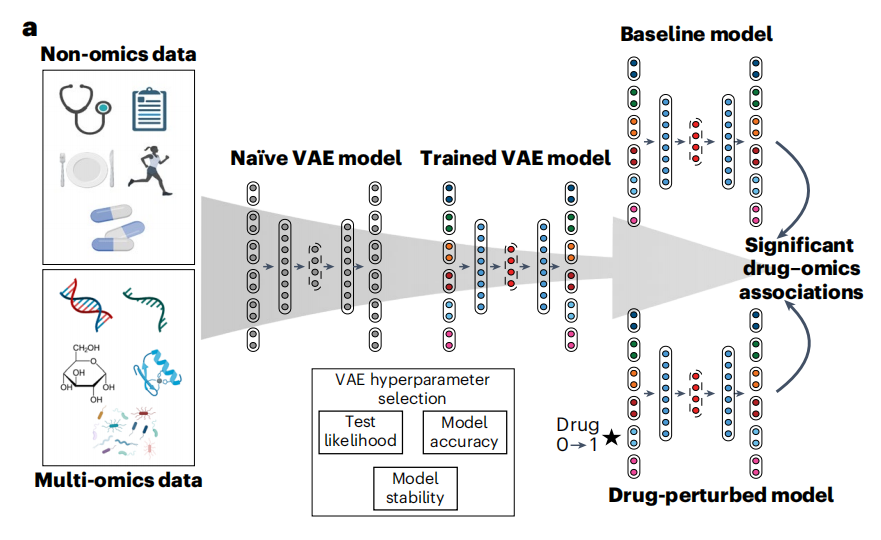
图:MOVE的综合原理与运用分析方法
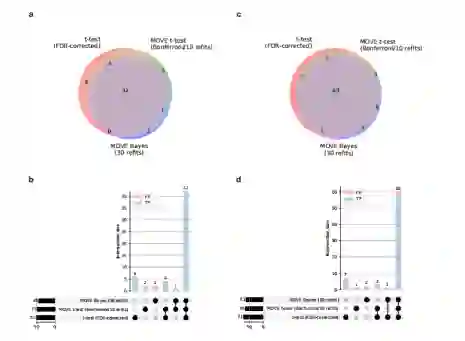
由于VAE模型是随机的,作者使用了一系列模型的结果,并开发了两种不同的方法来识别显著的关联。一种方法是基于在四个不同的模型上应用带有Bonferroni校正的t检验,其中每个模型都被改装10次(MOVE t-test),而作者也受到早期变分工作的启发,也使用了Bayes决策理论并且对单一模型改装30次(MOVE Bayes)。为了确定方法的不同参数,以便在标准方法之间进行比较(t-检验,方差分析(ANOVA)),作者将它们应用于由随机临床、药物和多组学数据组成的两个数据集。作者的研究结果表明,与t检验和方差分析相比,MOVE t检验和MOVE Bayes在识别药物组学关联方面表现良好,真实错误发现率(FDR)为0.05。
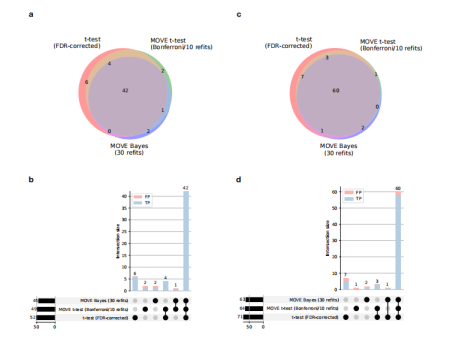
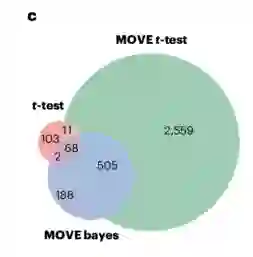
图:两个随机数据集方法的重叠
确定药物和多组学的关联 作者应用MOVE框架来识别DIRECT多模态数据中的药物关联。MOVE t-检验和MOVE Bayes两种方法分别识别出3,143个和763个与多组学和临床特征显著相关。作者分析了两种方法的交集,发现两种方法都发现了763个显著相关性中的573个(75%)。
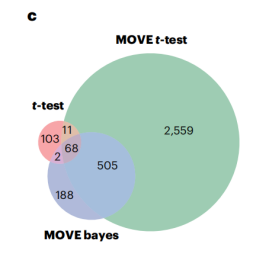
图:输入数据的标准t检验、MOVE t检验和MOVE Bayes方法之间药物组学相关性显著重叠
作为保守选择,作者使用两种方法确定的相关性进行进一步分析,与传统的检验(如t-检验和方差分析)相比,作者发现从184到573增加了211%的显著关联。
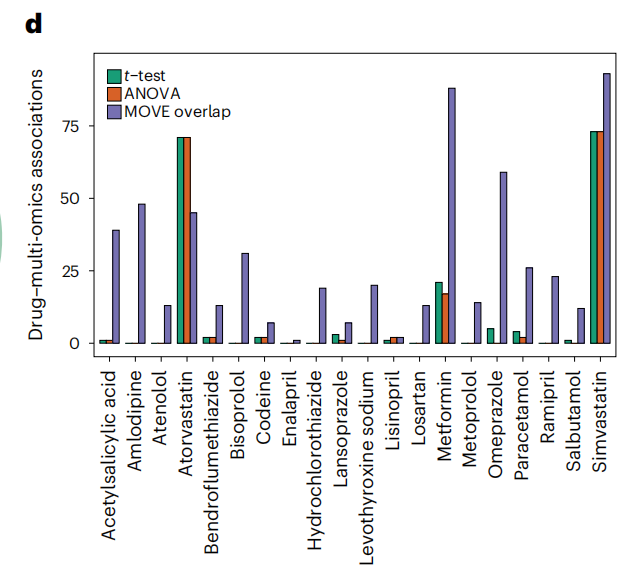
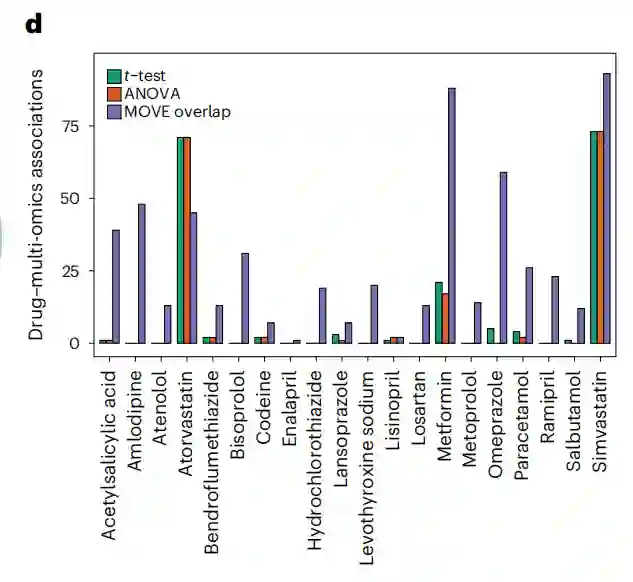
图:使用MOVE t-test与MOVE Bayes(紫色)、t-test(绿色)与方差分析(橙色)的中药物与特征显著关联的数量
此外,MOVE发现的显著相关性分布在所有药物中(双侧t检验,P = 0.016),而不仅仅是对大多数人使用的药物,如辛伐他汀、阿托伐他汀和二甲双胍。例如,MOVE确定每种药物的关联中位数为20,而t检验为1,方差分析为0,突出表明作者的方法对提取给予较少个体的药物关联更敏感。在多组学数据集中,作者发现最大数量的显著药物关联是与代谢组学、临床、以及转录组数据,每种药物平均有6个关联。结果表明,与传统的t检验和ANOVA相比,MOVE t检验和MOVE Bayes在识别药物-组学关联方面表现更佳。
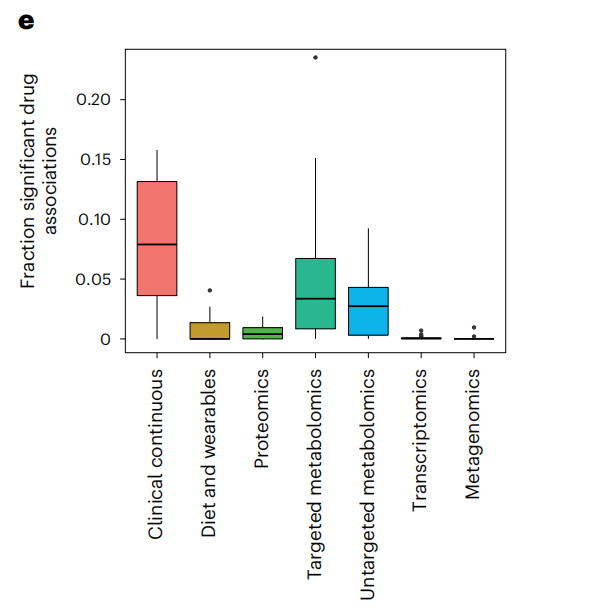
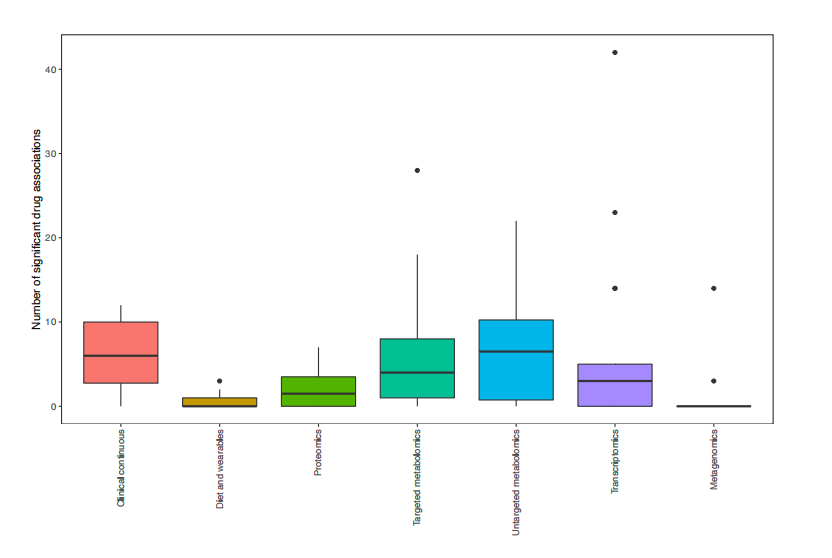
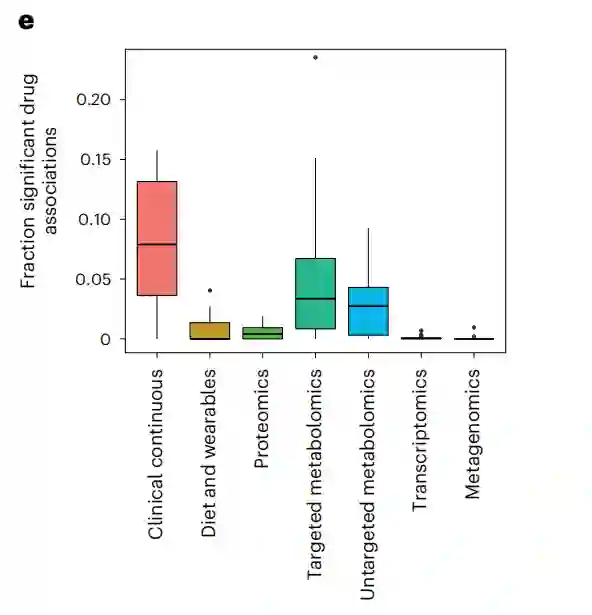
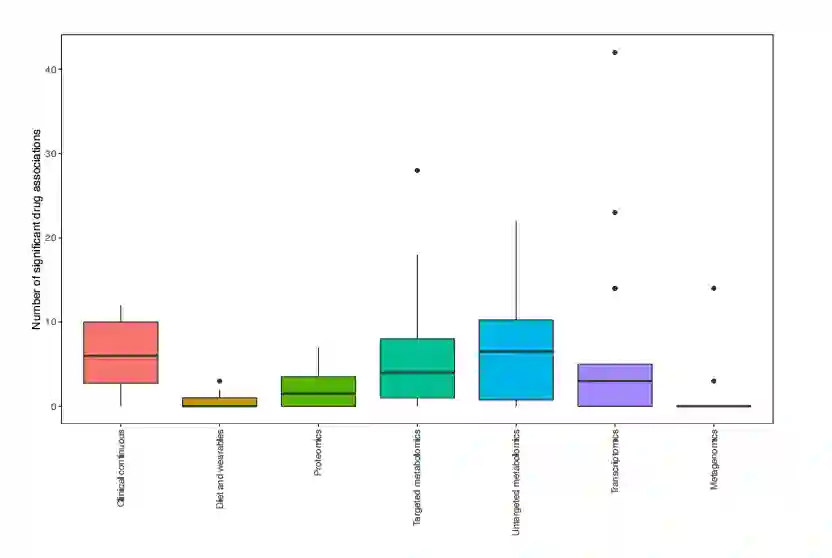
图:多组学数据集中特征与至少一种药物显著相关的比例(n = 20)(左)与有效关联的绝对数量(右)
当对所有可能的关联进行归一化时,与临床数据相关的关联比例最高(8%),其次是靶向代谢组学和非靶向代谢组学,与药物相关的特征平均分别为5.1%和2.8%。作者使用了Wesolowska-Andersen和Brorsson等人基于32个临床特征的聚类的四个原型聚类来调查结果是否可以由T2D队列中的疾病亚型驱动。最终,结果发现中位数为6.5%的显著药物组学关联是特定于一个亚组的,表明这些关联主要不是由原型驱动的。
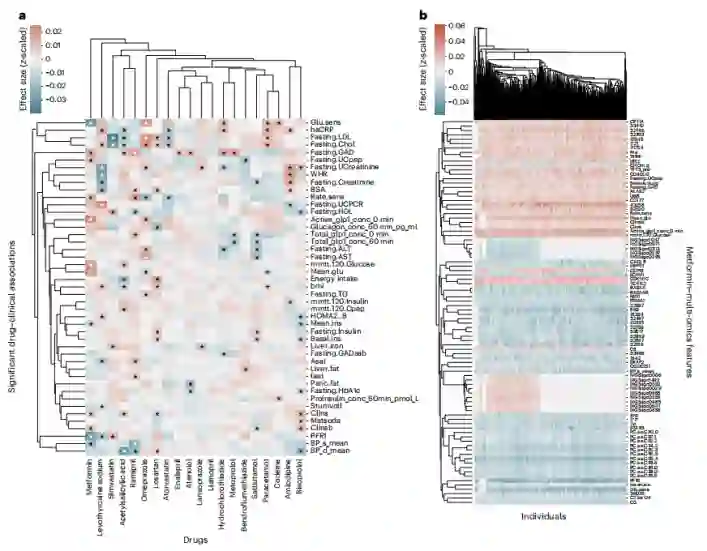
T2D生物标志物的变化与二甲双胍有关 作者调查了药物和多组学的相互作用,并最初关注预期的临床药物相互作用。例如,对于二甲双胍,作者在所有数据集中确定了88个重要的临床和多组学相互作用。在调查个体间的关联时,作者发现患者内部的低变异性表明变化是稳定的。经过试验,作者发现二甲双胍与胰岛素清除率、GLP-1活性、混餐糖耐量试验血糖水平、葡萄糖敏感性和血压等12项T2D临床标志物显著相关。
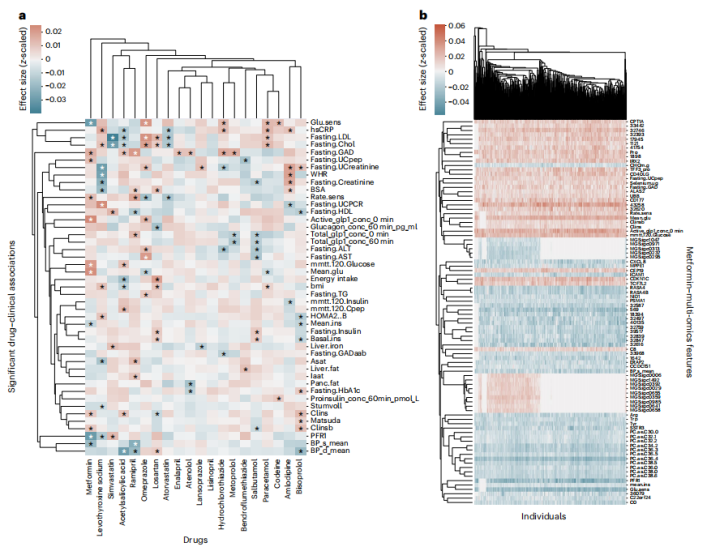
图:药物、临床和多组学特征之间的显著相关性
由于所有个体都有T2D,所以糖尿病状态的混杂效应不能从二甲双胍的影响中分离出来。在研究二甲双胍的多组学相关性时,作者发现7个相关蛋白中的2个(ERAP2和CD40L)可能与免疫系统相关。
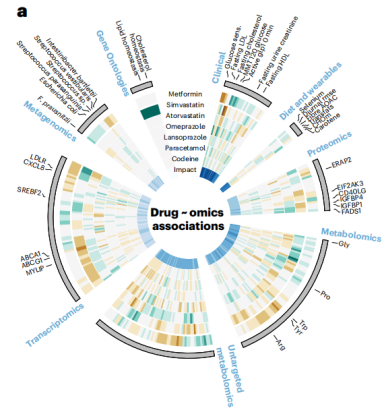
图:显示(从外到内)二甲双胍、辛伐他汀、阿托伐他汀、奥美拉唑、兰索拉唑、扑热息痛和可待因的效果大小(z刻度单位)
同样,在转录组学数据中,作者发现CXCL8和CD177被二甲双胍改变,而前者在健康个体和癌症患者中被证明发生了改变。在目标代谢组学数据中,作者发现了与氨基酰基- tRNA生物合成相关的代谢物的显著富集(超几何检验,P = 2.2 × 10−4,FDR校正)。最后,对于非靶向代谢组学数据,二甲双胍在所有药物中具有最高的关联数(22个关联),这表明二甲双胍治疗的新的代谢效应可能被识别出来。
二甲双胍和奥美拉唑与肠道菌群的关系 最近的研究表明,药物摄入影响人体肠道微生物组的组成。在此,作者发现二甲双胍和奥美拉唑是唯一与宏基因组数据有显著关联的药物,增加了11个宏基因组物种,而减少了6个其他物种。

图:与二甲双胍显著相关的人类肠道宏基因组物种的效应量
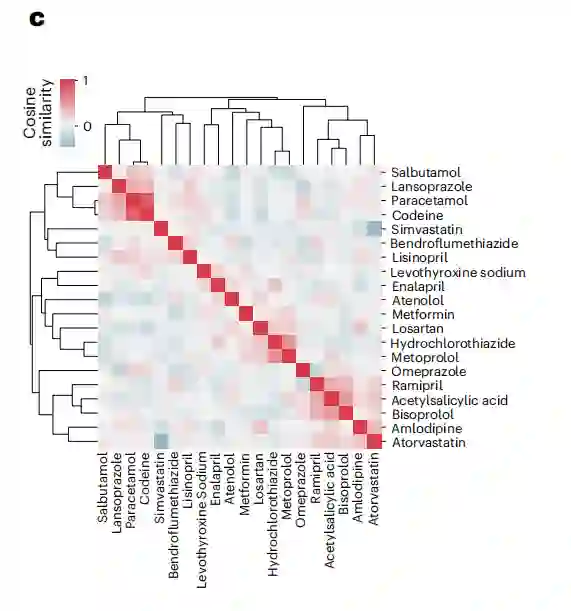
多组学数据的药物多药性和相似性 作者调查了药物及其多组学关联之间的相似性。总的来说,作者观察到四个簇,每个簇包含3到6种药物,并发现簇中的一些药物可能与多药作用有关。

图:通过比较多组学数据集的药物反应概况以确定药物-药物相似性
作者研究了药物-药物组合对相关性的影响,发现总体药物关联相似性与服用两种药物的个体之间存在相关性(PCC 0.75, P值为2.2 × 10−35)。这一发现表明,同时服用两种药物可能会产生多药效应,从而在所有临床和多组学变化中产生更高的药物-药物相似性。此外,具有最相似的药物和多组学关联的药物是可待因和扑热息痛,余弦相似度为0.78。
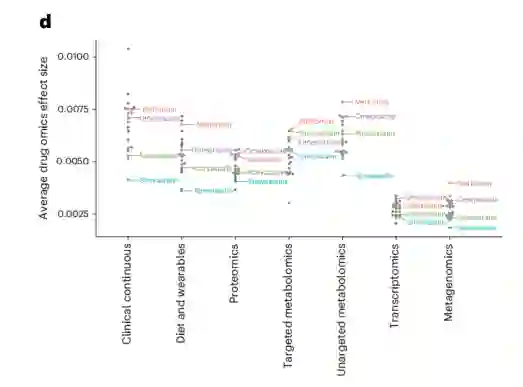
药物的影响在组学数据中广泛存在 作者发现肠道微生物组是所有药物中具有统计学意义的命中次数第二少的数据集,有17个显著关联。只有饮食和可穿戴数据的相关性较低(11);转录组学、蛋白质组学、靶向代谢组学和非靶向代谢组学有44-134个显著相关性。然后,作者询问了不同数据集中药物的效应量是否不同,并确定了各自多组学数据集中药物的累积效应量。在这里,作者发现转录组和宏基因组数据中的平均效应值是所有药物中最低的,而宏基因组数据集中的平均效应值显著低于除转录组外的所有其他组学数据集(ANOVA, Tukey HSD检验,校正后P < 0.05)。
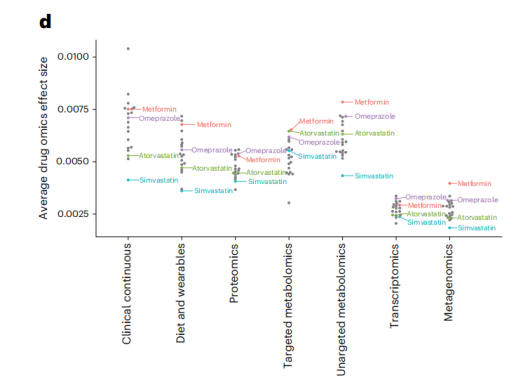
图:药物对组学数据集的平均效果(z-score)
当作者将亚组分析显著的药物组学关联时,其中肠道微生物组中只有两种药物具有显著关联(二甲双胍和奥美拉唑),作者发现,与其他多组学数据集的效应量相比,这两种药物的效果相似或更低。这一观察结果表明,多组学对药物刺激的反应不仅针对肠道微生物组,而且说明试图了解药物作用时应包括多组学数据集。
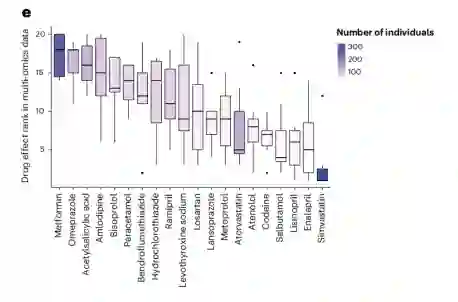
多组学数据中对药物的影响进行排名 最后,作者在多组学数据集中调查了单个药物的效应量。作者发现,二甲双胍和奥美拉唑对多组学数据(累积等级评分)的影响最为显著,两种他汀类药物在20种药物中排名第14和第20位,而辛伐他汀的累积效应值总体排名最低。这一分析并没有被服用特定药物的个体数量所混淆,因为个体数量与药物效果之间没有相关性(PCC = 0.14)。这与仅调查他汀类药物排名第2和第4且效应值高的显著相关性时相反。这一观察结果可能表明他汀类药物的强效较少,然而,平均排名最高的二甲双胍和奥美拉唑都有较大的系统效应。
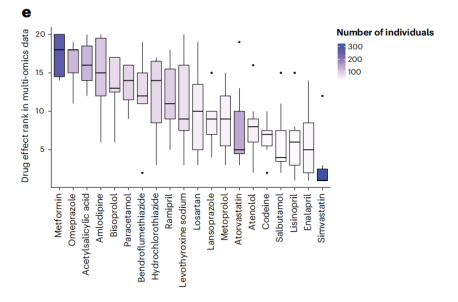
图:不同药物的多组学分布排序。
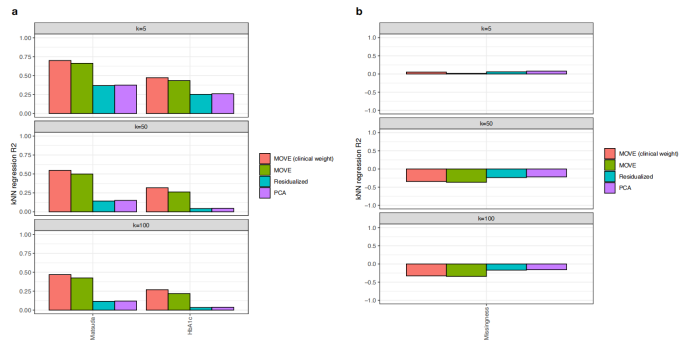
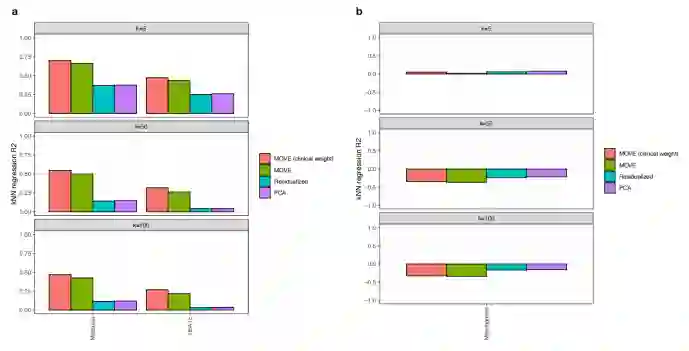
图:kNN回归显示MOVE可以识别临床相关的模式
结论
作者展示了使用无监督深度学习来整合和提取深度表型的T2D人群的关联是可能的,通过使用VAE模型的生成能力进一步实现了这一目标。与传统的单变量统计检验相比,MOVE可以为更广泛的药物选择识别显著的药物组学关联。作者通过添加了19种额外药物的用药数据,并将所有数据作为无监督深度学习模型的输入,允许模型同时从所有输入中学习,因此能够确定包括二甲双胍在内药物和多组学数据之间的关联,表明垂直整合的重要性。
作者强调,该方法不限于药物协会。原则上,所有组学数据都可以评估数据集之间的关联。因此,作者相信,该生成方法为大型多组学数据分析提供了新的可能性,可以发现潜在的新生物标志物,进行思维实验并在高维分子数据中调查药物的潜在直接影响,从而得出可测试的假设。 参考资料 Allesøe, R.L., Lundgaard, A.T., Hernández Medina, R. et al. Discovery of drug–omics associations in type 2 diabetes with generative deep-learning models. Nat Biotechnol (2023). https://doi.org/10.1038/s41587-022-01520-x