
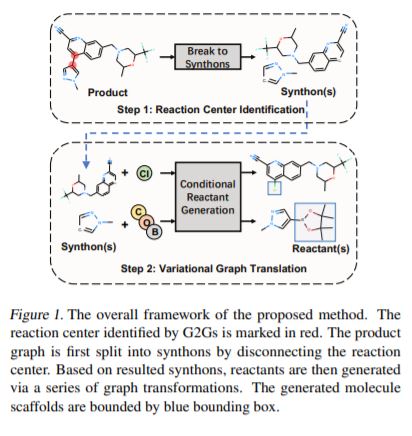
新药发现(Drug Discovery)领域中的一个基础问题是预测目标分子的合成路线,即逆合成预测(Retrosynthesis Prediction)。现有的方法大多将给定的目标分子(产物)与许多化学反应模版匹配,从而预测可能的反应物。然而,模版匹配耗费大量的算力,并且这些方法在新数据集上的泛化能力也欠佳。本文提出了一种名为G2Gs的不依赖化学反应模版的方法。G2Gs通过一系列图变换,将产物分子转换(或称为翻译)到反应物分子。G2Gs首先通过一个反应中心预测模块,将产物分子分解为多个合成子。然后它通过一个变分图翻译模块,将每个合成子转换到最终的反应物分子。实验结果表明,本文提出的方法的性能远优于那些不依赖反应模版的方法。并且,G2Gs的性能与基于模版的方法相近,但它不依赖任何领域知识,也有更好的可扩展性。
本文第一作者史晨策是北大计算机科学技术系2016级本科生,也是第一届图灵班学生,获得北京大学信息科学技术学院“十佳”优秀本科毕业论文奖,已被MILA唐建教授录为研究生。
成为VIP会员查看完整内容
相关内容
专知会员服务
12+阅读 · 2020年4月1日
专知会员服务
136+阅读 · 2020年3月8日
Arxiv
10+阅读 · 2020年4月9日
Arxiv
7+阅读 · 2018年5月25日
相关VIP内容
专知会员服务
12+阅读 · 2020年4月1日
专知会员服务
136+阅读 · 2020年3月8日
相关资讯
相关论文
Arxiv
10+阅读 · 2020年4月9日
Arxiv
7+阅读 · 2018年5月25日