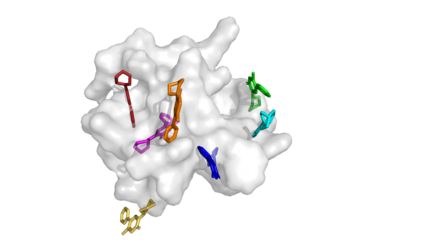
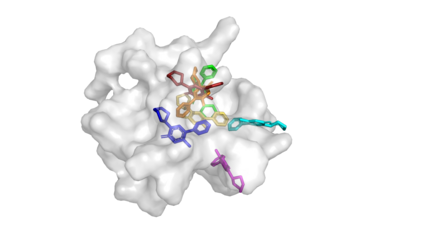
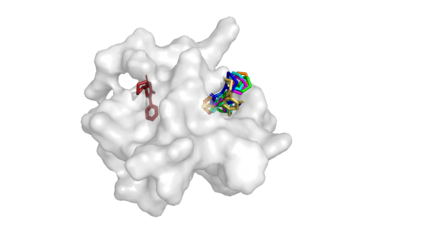
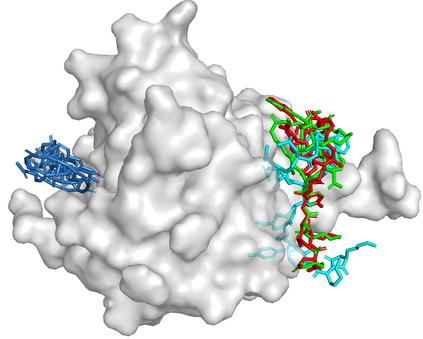
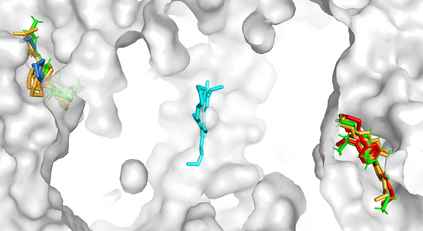
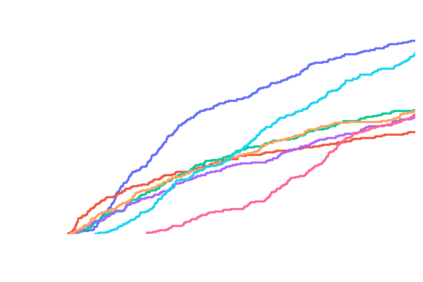
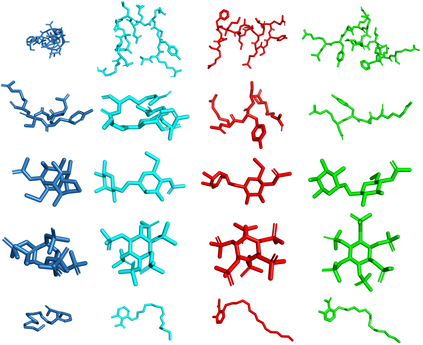
Predicting the binding structure of a small molecule ligand to a protein -- a task known as molecular docking -- is critical to drug design. Recent deep learning methods that treat docking as a regression problem have decreased runtime compared to traditional search-based methods but have yet to offer substantial improvements in accuracy. We instead frame molecular docking as a generative modeling problem and develop DiffDock, a diffusion generative model over the non-Euclidean manifold of ligand poses. To do so, we map this manifold to the product space of the degrees of freedom (translational, rotational, and torsional) involved in docking and develop an efficient diffusion process on this space. Empirically, DiffDock obtains a 38% top-1 success rate (RMSD<2A) on PDBBind, significantly outperforming the previous state-of-the-art of traditional docking (23%) and deep learning (20%) methods. Moreover, DiffDock has fast inference times and provides confidence estimates with high selective accuracy.
翻译:将小分子悬浮对蛋白 -- -- 称为分子对齐的任务 -- -- 的绑定结构预测成一个小分子对蛋白 -- -- 这对于药物设计至关重要。最近将对接作为回归问题的深层学习方法比传统的基于搜索的方法减少了运行时间,但比起传统的搜索方法,还不能大幅度提高准确性。我们把分子对接作为基因模型问题来设置,并开发DiffDock,这是比起非欧洲的悬浮成形体的分散基因化模型。为了做到这一点,我们绘制了这个元件,以产品空间的自由度(转换、旋转和旋转)空间为产品空间。DiffDock在对接和开发这一空间的有效扩散过程方面,获得了38%的最高一级成功率(RMSD < 2A),大大超过了以前传统的对接(23 % ) 和深层次学习(20%) 的方法。此外,DiffDock具有快速的推算时间,并以高选择性的精确度提供了信心估计。