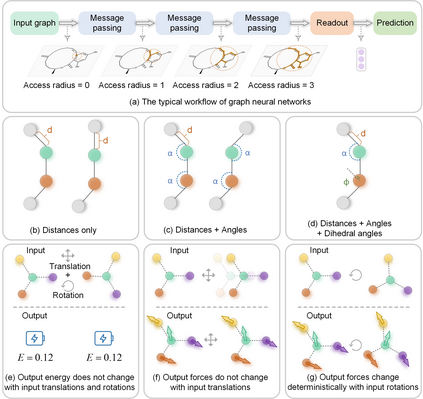
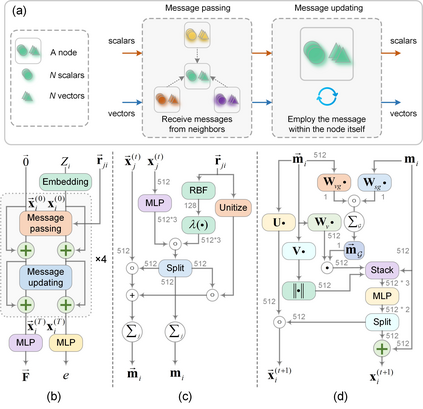
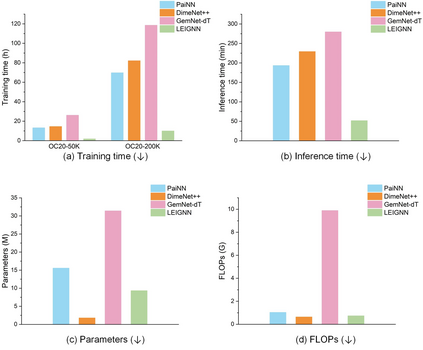
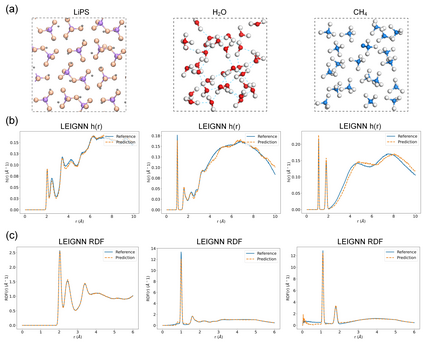
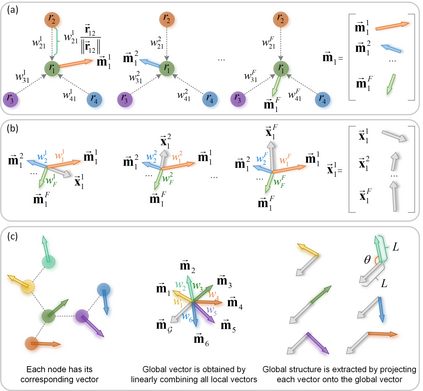
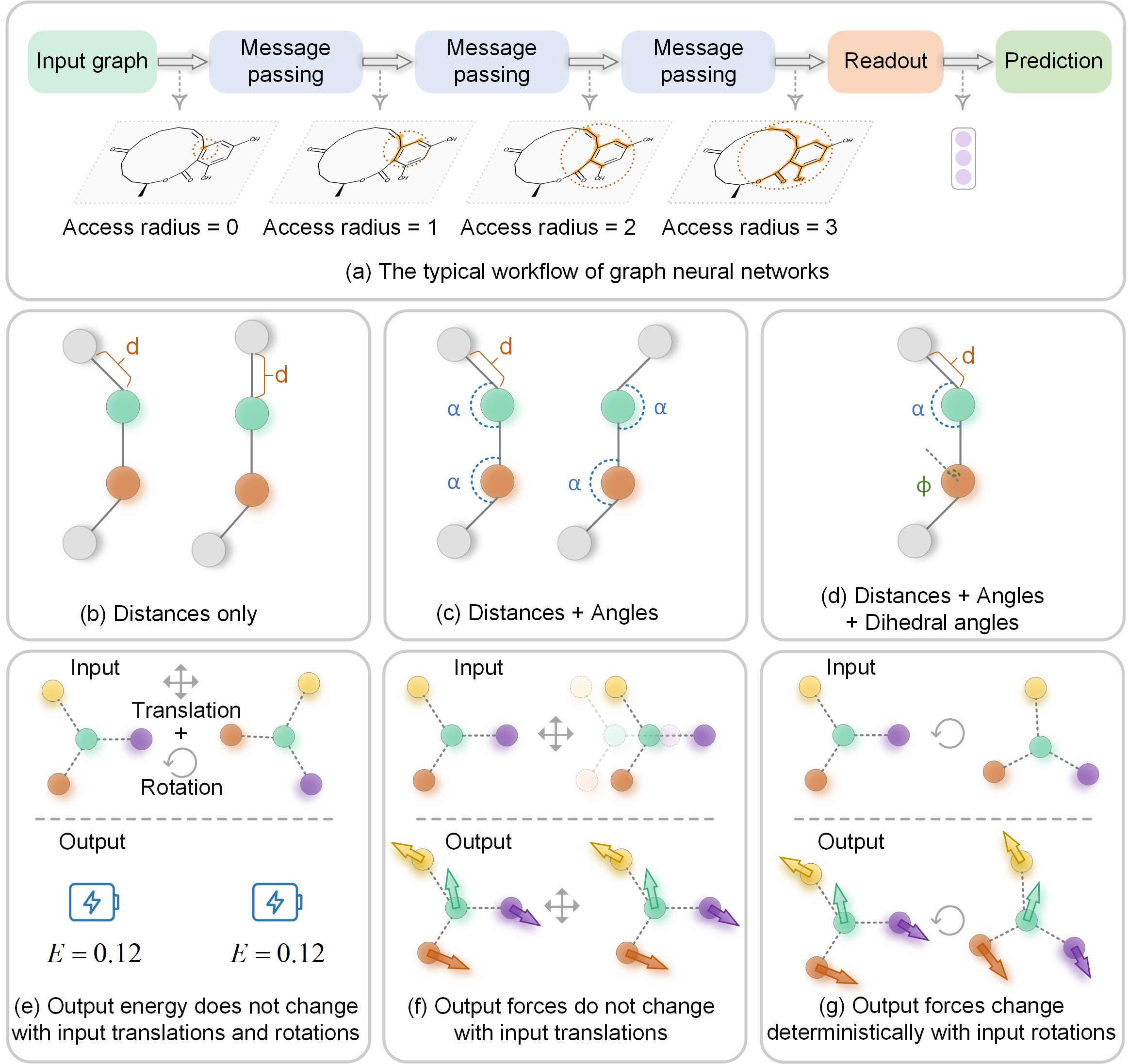
In modern computational materials science, deep learning has shown the capability to predict interatomic potentials, thereby supporting and accelerating conventional simulations. However, existing models typically sacrifice either accuracy or efficiency. Moreover, lightweight models are highly demanded for offering simulating systems on a considerably larger scale at reduced computational costs. A century ago, Felix Bloch demonstrated how leveraging the equivariance of the translation operation on a crystal lattice (with geometric symmetry) could significantly reduce the computational cost of determining wavefunctions and accurately calculate material properties. Here, we introduce a lightweight equivariant interaction graph neural network (LEIGNN) that can enable accurate and efficient interatomic potential and force predictions in crystals. Rather than relying on higher-order representations, LEIGNN employs a scalar-vector dual representation to encode equivariant features. By extracting both local and global structures from vector representations and learning geometric symmetry information, our model remains lightweight while ensuring prediction accuracy and robustness through the equivariance. Our results show that LEIGNN consistently outperforms the prediction performance of the representative baselines and achieves significant efficiency across diverse datasets, which include catalysts, molecules, and organic isomers. Finally, to further validate the predicted interatomic potentials from our model, we conduct classical molecular dynamics (MD) and ab initio MD simulation across various systems, including solid, liquid, and gas. It is found that LEIGNN can achieve the accuracy of ab initio MD and retain the computational efficiency of classical MD across all examined systems, demonstrating its accuracy, efficiency, and universality.
翻译:暂无翻译