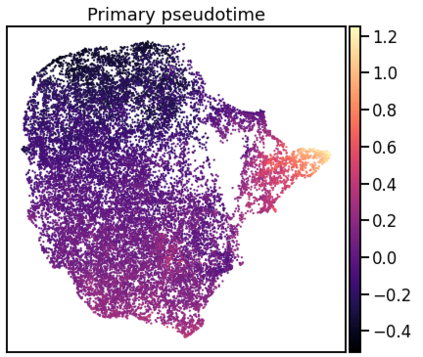
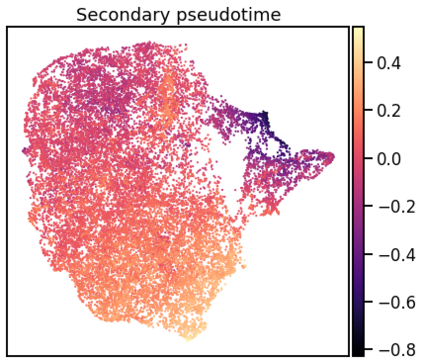
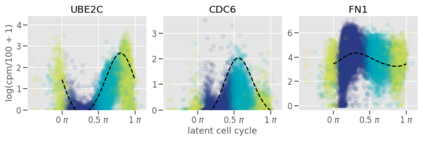
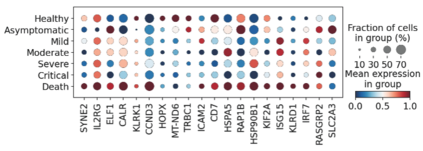
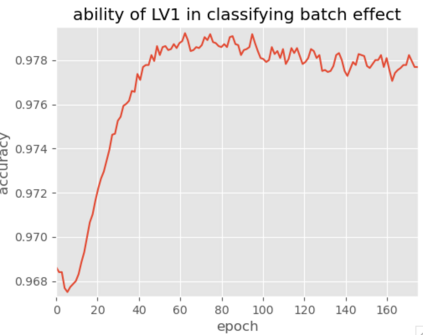
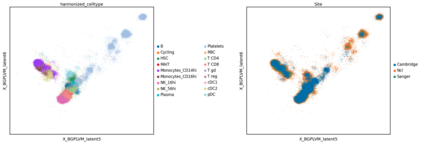
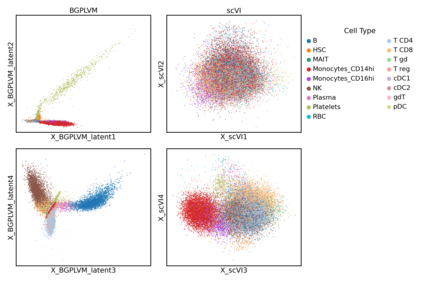
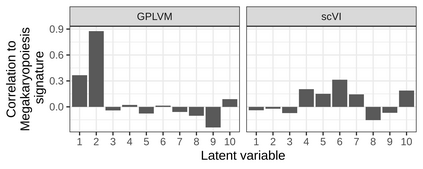
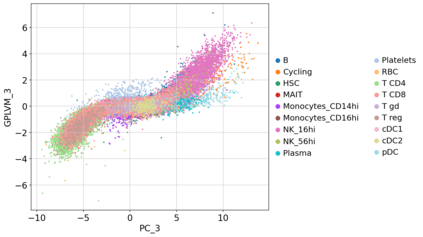
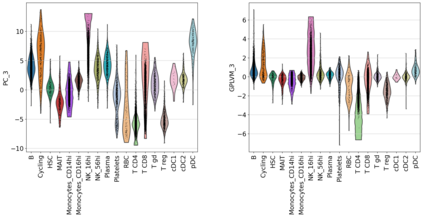
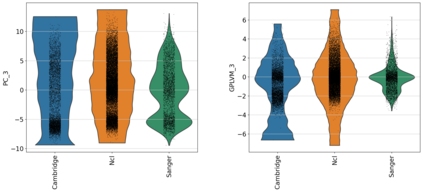
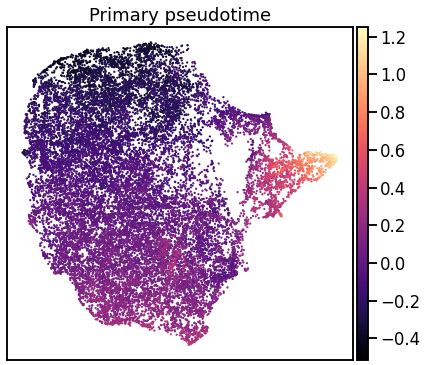
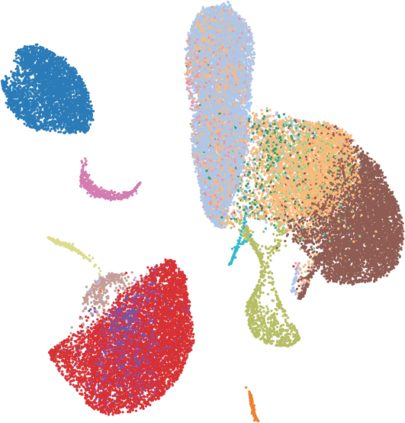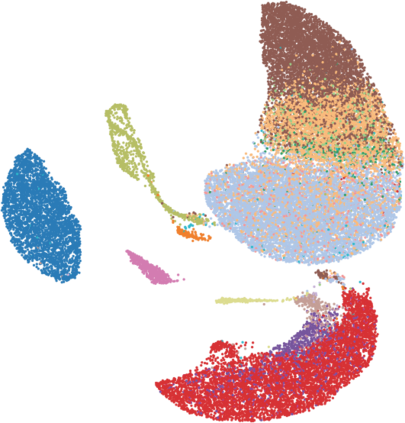Single-cell RNA-seq datasets are growing in size and complexity, enabling the study of cellular composition changes in various biological/clinical contexts. Scalable dimensionality reduction techniques are in need to disentangle biological variation in them, while accounting for technical and biological confounders. In this work, we extend a popular approach for probabilistic non-linear dimensionality reduction, the Gaussian process latent variable model, to scale to massive single-cell datasets while explicitly accounting for technical and biological confounders. The key idea is to use an augmented kernel which preserves the factorisability of the lower bound allowing for fast stochastic variational inference. We demonstrate its ability to reconstruct latent signatures of innate immunity recovered in Kumasaka et al. (2021) with 9x lower training time. We further analyze a COVID dataset and demonstrate across a cohort of 130 individuals, that this framework enables data integration while capturing interpretable signatures of infection. Specifically, we explore COVID severity as a latent dimension to refine patient stratification and capture disease-specific gene expression.
翻译:单细胞RNA-seq数据集的大小和复杂性都在增加,使得能够研究各种生物/临床环境中细胞构成的变化。可缩放的维度减少技术需要分解这些技术的生物变异,同时考虑技术和生物混淆者。在这项工作中,我们推广了一种普及的概率非线性非线性减少方法,高森过程潜伏变量模型,以扩大为大规模的单细胞数据集,同时明确计算技术和生物混淆者。关键思想是使用一个强化的内核,以保持允许快速随机变异推断的低约束系数。我们展示其重建在Kumasaka等人(2021年)恢复的内核免疫潜在特征的能力,同时减少9x培训时间。我们进一步分析COVID数据集,并在130人组中显示,这一框架能够在获取可解释的感染特征的同时进行数据整合。具体地说,我们探索COVID严重程度作为潜在层面,以完善病人的分层和捕捉到特定疾病的基因表达方式。