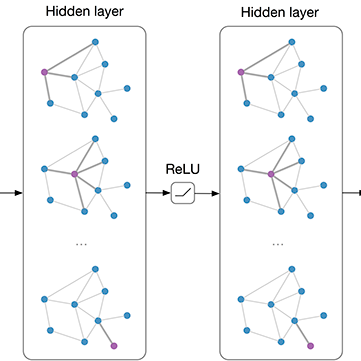
Early detection of cancer plays a key role in improving survival rates, but identifying reliable biomarkers from RNA-seq data is still a major challenge. The data are high-dimensional, and conventional statistical methods often fail to capture the complex relationships between genes. In this study, we introduce RGE-GCN (Recursive Gene Elimination with Graph Convolutional Networks), a framework that combines feature selection and classification in a single pipeline. Our approach builds a graph from gene expression profiles, uses a Graph Convolutional Network to classify cancer versus normal samples, and applies Integrated Gradients to highlight the most informative genes. By recursively removing less relevant genes, the model converges to a compact set of biomarkers that are both interpretable and predictive. We evaluated RGE-GCN on synthetic data as well as real-world RNA-seq cohorts of lung, kidney, and cervical cancers. Across all datasets, the method consistently achieved higher accuracy and F1-scores than standard tools such as DESeq2, edgeR, and limma-voom. Importantly, the selected genes aligned with well-known cancer pathways including PI3K-AKT, MAPK, SUMOylation, and immune regulation. These results suggest that RGE-GCN shows promise as a generalizable approach for RNA-seq based early cancer detection and biomarker discovery (https://rce-gcn.streamlit.app/ ).
翻译:癌症的早期检测对提高生存率至关重要,但从RNA-seq数据中识别可靠的生物标志物仍是一项重大挑战。这些数据具有高维度特性,传统统计方法往往难以捕捉基因间的复杂关系。本研究提出了RGE-GCN(基于图卷积网络的递归基因消除框架),该框架将特征选择与分类整合于单一流程中。我们的方法从基因表达谱构建图结构,利用图卷积网络对癌症与正常样本进行分类,并应用积分梯度法突出最具信息量的基因。通过递归剔除相关性较低的基因,模型最终收敛于一组兼具可解释性与预测性的紧凑生物标志物集合。我们在合成数据以及肺癌、肾癌和宫颈癌的真实RNA-seq队列中对RGE-GCN进行了评估。在所有数据集中,该方法均持续取得比DESeq2、edgeR和limma-voom等标准工具更高的准确率与F1分数。值得注意的是,筛选出的基因与PI3K-AKT、MAPK、SUMO化修饰及免疫调控等已知癌症通路高度吻合。这些结果表明,RGE-GCN有望成为基于RNA-seq的早期癌症检测与生物标志物发现的通用化方法(https://rce-gcn.streamlit.app/)。