AI研发新药真有那么神?可能哈佛、斯坦福和阿斯利康实验室都在吹牛
安妮 李林 编译自 Medium
量子位 出品 | 公众号 QbitAI

近年来,向往着用AI研发新药的美好愿景,巨头纷纷投下了重注。
制药巨头赛诺菲和AI药物发现平台Exscientia签下3亿美元巨额订单,葛兰素史克随后也出了4200万美元。默克和Atomwise、强生旗下的杨森制药与BenevolentAI也都有类似的合作。
风投也非常看好这个领域。硅谷VC公司安德森·霍洛维茨(A16Z)为投资AI药物发现公司,组建了一支4.5亿美元的基金。
好像巨头都上车了,小玩家要不要跟上?
今天,硅谷极客们最爱的HackerNews上有一篇热门文章就在探讨这件事。大部分评论者都同意这篇文章的基本观点:AI在药物发现中的作用被过度吹捧了。
这篇文章通过分析制药公司阿斯利康、名校哈佛和斯坦福、以及创业公司Insilico Medicine等名声卓著的实验室最近的一些研究,来说明AI研究者是如何过度吹捧他们的成果的。
以下是这篇热门文章的主要内容,量子位编译整理:

在默克,用计算机设计药物
我乐观地认为,这些事情2018年会有变化。这可能并不是因为人工智能的突破,而是因为这些研发机构会有进步:现在,随着在线教育和社交媒体的兴起,更强力的检查和平衡已经成为可能,为开放同行评议提供了新机会,这会成为挤泡沫的力量。
接下来,让我们进入技术部分,谈谈最近一些过度吹捧的AI研究案例。
阿斯利康

最近,英国制药公司阿斯利康和明斯特大学、上海大学的研究者一起发表了一项研究,尝试用循环神经网络(RNN)和强化学习来生成新分子。
相关论文:
Generating Focussed Molecule Libraries for Drug Discovery with Recurrent Neural Networks
https://arxiv.org/abs/1701.01329
这个问题很重要,因为一个创造性的AI能为发现引导物的流程中带来更多多样性。
这篇论文引起了我的注意,是因为用了很大篇幅来讲模型的评价。这看起来很有深度,他们给出了基于谷本系数相似性度量和基于levenshtein距离等多个衡量指标。论文中还有大量的可视化展示。
然而,他们的衡量标准都是用在AI生成的分子与自然分子之间的,总是“省略”了对AI产生的分子彼此之间距离的测量。这种省略让人有一种“多样性”的错觉:AI生成的分子和自然分子之间的距离很大,就可以认为AI具有创造性,探索了化学空间中的新方向。
于是我们就有了如下图所示的错觉:
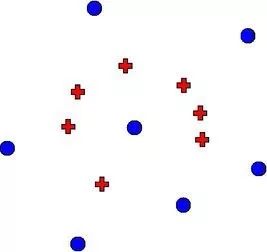
△ 真正的多样性:蓝色圆点代表AI生成的分子,红色十字代表自然分子
而实际上,如果AI生成的分子之间距离很小,这就意味着我们陷入了下图所示的状况,模型所产生的分子基本位于同一位置,并没有多样性:
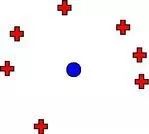
△ 多样性错觉:AI生成的分子(蓝色圆点)和自然分子(红色十字)之间仍然有很大距离,但AI生成的分子集中在一起
简单来说,阿斯利康这篇论文对房间里的大象视而不见,最近这家公司一些其他论文依然有这个问题。
比如:
Molecular De Novo Design through Deep Reinforcement Learning
https://arxiv.org/abs/1704.07555
Application of generative autoencoder in de novo molecular design
https://arxiv.org/abs/1711.07839

哈佛

哈佛的一个团队注意到了这个多样性的问题。他们通过查看人工智能生成的样本,感觉出了问题,想要做点什么,于是提出了ORGAN模型。
相关论文:
Objective-Reinforced Generative Adversarial Networks (ORGAN) for Sequence Generation Models
https://arxiv.org/abs/1705.10843
Optimizing distributions over molecular space. An Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry (ORGANIC)
https://chemrxiv.org/articles/ORGANIC_1_pdf/5309668/3
GitHub地址:
https://github.com/gablg1/ORGAN
他们的想法是要带来更丰富的化学多样性和化学现实主义,用第二个神经网络——鉴别器来校正生成器。如果分子看起来不太自然,它就惩罚生成器。这个想法来自AI学术界火爆的“生成对抗网络(GAN)”。
这是个好想法,但执行得非常糟糕。他们的结论是ORGAN比较好,但这个说法只是基于他们自己的观察得到的,没有任何量化支持。他们的定量实验并不支持这一结论。
而且,他们训练模型的方式是有问题的。看他们在GitHub上公开的训练日志就会发现,鉴别器总是在高度惩罚生成器,这个鉴别器太追求完美了,根本就是消灭了GAN的好处。
斯坦福

斯坦福有个用AI和深度学习研究化学的大团队,团队负责人是Vijay Pande,具有斯坦福教授和A16Z风投公司投资人的双重身份。
在Vijay Pande管理的投资项目中,“专为用机器学习方法测试分子性质而设计的基准”MoleculeNet最受关注的一个。
这个项目看起来画风严肃,包含大量化学知识、图形及深度学习模型,尤其关注graph-CNN和斯坦福团队其他为化学问题特制的神经网络。
然而,还有一个大家心知肚明但却避而不谈的问题:上述Pande团队没有努力将他们的数据插入到一个字符级的卷积神经网络(char-CNN)中。自2015年Yann LeCun的论文Text Understanding from Scratch提出这种方法后,字符级CNN就容易被例行公事地用在各种AI任务中进行文本处理,并且比graph-CNN简单得多。
如果想使用字符级CNN,插入“SMILES(simplified molecular-input line-entry system)”字符串就好了。
回过头来想想,为什么斯坦福的研究人员不选择这种更简单的操作呢?在论文MoleculeNet: A Benchmark for Molecular Machine Learning的第17页,我们可以看到:
最近的研究已经证明了用更复杂的方法从SMILES字符串学习有用表示的能力,所以在不久的将来,将SMILES字符串用于进一步的学习任务是可行的。
MoleculeNet: A Benchmark for Molecular Machine Learning
https://arxiv.org/abs/1703.00564
我不太相信字符级CNN对这个斯坦福团队来说过于复杂了,他们甚至在另外一篇文章中用到了字符级CNN。
Retrosynthetic reaction prediction using neural sequence-to-sequence models
https://arxiv.org/abs/1706.01643
一个更合理、更尴尬的解释是,他们害怕字符级CNN的效果会更好。这意味着,他们心爱的graph-CNN模型将在分子基准测试中被击败,而这并不符合他们的期望。
原因很简单。DeepChem是一个斯坦福领导的开源库,用来实现MoleculeNet模型。如果字符级CNN比graph-CNN效果好,那么实践者就无需DeepChem了,他们可以直接用简单的TensorFlow或Pytorch实现。
在2018年,开源框架是一种战略性资产,就像通过Android,谷歌主宰了移动操作系统市场一样。DeepChem可能也想走一条类似的道路,控制AI在药物发现领域的发展。这可能就是MoleculeNet对字符级CNN视而不见的原因。
我自己使用DeepChem的经历印证了这个推测。曾经我天真地想在项目中使用DeepChem,直到我发现,无法将DeepChem模型和非DeepChem模型混合在一起。用DeepChem辨别器和非DeepChem生成器对对抗训练很有用,但我无法实现这个想法。相反,我被牢牢锁在DeepChem的代码中,没有料想到还有如此恶意的东西。
为了逃离这个陷阱,让DeepChem真正开源,我不得不挖掘复杂的代码。
项目地址:
https://github.com/mostafachatillon/deepchem
对于一个更成熟的项目来说,这样做会更加困难。所以我的印象是,用这种“厂商锁定”的策略,DeepChem想吃掉AI世界来做化学研究。所以我对他们的合作投资人中有Marc Andreessen一点都不惊异。

虽然MoleculeNet的团队成员刻意避免用字符级CNN做基准测试,但他们仍为MoleculeNet和DeepChem设计了精美的网页。这表明他们还是太注重表面,PR优先于坚实的科学研究。这也是硅谷的典型策略,初创公司设计虚假产品吸引流量,然后依靠社区来构建真实的东西。

Insilico Medicine

Insilico Medicine是AI创业公司中生成模型的先驱。在一篇论文中,Alex Zhavoronkov和他的团队提出了DruGAN,一种先进的生成式对抗自编码模型。我一直搞不明白这个模型哪里先进了。
druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico
http://pubs.acs.org/doi/abs/10.1021/acs.molpharmaceut.7b00346
就发现药物这个需求来说,DruGAN肯定已经不先进了:它和其他的生成模型有相同的缺点,可能会让人失望。
在最近的一篇论文中他们使用了更复杂的工具,但也称不上先进吧。在他们论文的第9-10页,有这样一段话:
本研究使用的MACCS分子指纹不是理想的分子结构表征。Direct SMILES、InChI、分子图以及其他化学和生物相关的分子结构表征可作为更好的训练类型。
他们甚至还没有用到基准测试的变分自编码器(VAE)。在论文中他们声称DruGAN比VAE好,但是在Github上,一个DruGAN的作者提出了相反的观点:
实际上,我们没有像AAE[DruGAN]那样调优VAE网络,所以这种比较不公平。我的意思是,你可以优化VAE,并超越我们的AAE。
所以我认为,DruGAN只比他们8个月前发表的论文先进。在整个论文中,他们不断提到相比以前工作的改进。也许,“进步”只是一个自我祝贺的词。
综上所述,我认为,AI在药物研发领域的应用被很多研究人员夸大了。
原文:https://medium.com/the-ai-lab/artificial-intelligence-in-drug-discovery-is-overhyped-examples-from-astrazeneca-harvard-315d69a7f863
— 完 —
加入社群
量子位AI社群13群开始招募啦,欢迎对AI感兴趣的同学,加小助手微信qbitbot5入群;
此外,量子位专业细分群(自动驾驶、CV、NLP、机器学习等)正在招募,面向正在从事相关领域的工程师及研究人员。
进群请加小助手微信号qbitbot5,并务必备注相应群的关键词~通过审核后我们将邀请进群。(专业群审核较严,敬请谅解)
诚挚招聘
量子位正在招募编辑/记者,工作地点在北京中关村。期待有才气、有热情的同学加入我们!相关细节,请在量子位公众号(QbitAI)对话界面,回复“招聘”两个字。

量子位 QbitAI · 头条号签约作者
վ'ᴗ' ի 追踪AI技术和产品新动态