NIPS'21 | 通过动态图评分匹配预测分子构象
编译 | 沈祥振
审稿 | 郭梦月

从 2D 分子图中预测稳定的 3D 构象一直是计算化学中的一个长期挑战。而最近,机器学习方法取得了相比传统的实验和基于物理的模拟方法更优异的成绩。这些方法主要侧重于模拟分子图上相邻原子之间的局部相互作用,而忽略了非键合原子之间的长程相互作用。然而,这些未成键的原子在 3D 空间中可能彼此接近,模拟它们的相互作用对于准确确定分子构象至关重要,尤其是对于大分子和多分子复合物。在本文中,作者提出了一种称为动态图评分匹配 (DGSM) 的分子构象预测新方法,该方法通过在训练和推理过程中根据原子之间的空间接近度动态构建原子之间的图结构来对局部和远程相互作用进行建模。具体来说,DGSM根据动态构建的图,使用评分匹配方法直接估计原子坐标对数密度的梯度场。可以以端到端的方式有效地训练整个框架。多项实验表明,DGSM 的表现远超该领域一流水平,并且能够为更广泛的化学系统生成构象,例如蛋白质和多分子复合物。
简介
基于图的分子表示普遍出现于各种任务之中,例如属性预测和分子生成。不过,分子更自然的表示方法是使用 3D 几何或 3D 构象,它将分子表示为一组 3D 坐标。分子的 3D 表示是许多任务的核心,例如分子性质预测和虚拟筛选。然而,确定分子的构象仍然是一项具有挑战性的任务——源于昂贵又耗时的计算方法(例如分子动力学 (MD))和实验方法(例如晶体学)。
最近,机器学习方法已被证明有望用于分子构象的生成。GRAPHDG 和 CGCF 等开创性的方法会先进行键合原子间距离的预测,并据此通过后处理算法求解 3D 坐标。最近, Shi 等人提出了 CONFGF ,它采用评分匹配技术来学习键合原子之间的伪力,并迭代地将这些力施加到随机初始化的 3D 结构上,直到收敛。CONFGF 突破了之前工作中的两段式方法,并显著提高了性能。然而,这些方法有一个共同的主要限制——它们主要侧重于模拟由输入分子图定义的键合原子之间的局部相互作用,但未能捕获非键合原子之间的长程相互作用,因为它们只根据键合原子之间的距离(或梯度)进行建模。
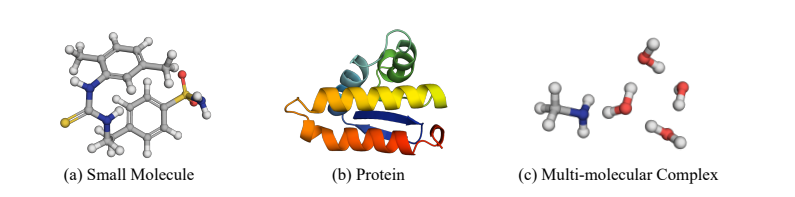
图1 三个分子系统,其中远程相互作用对其构象至关重要
而在分子力学中,改变构象的分子的势能可以建模为四部分的总和:
其中 Ebond、Eangle 和 Etorsion 对键合原子之间的局部相互作用进行建模,这些相互作用是在以前的方法中建模的。非键合原子之间的长程相互作用,表示为Enon-bonded,也是非平凡的,它通过不可忽略的静电力或范德华力等塑造分子几何形状。对于多分子复合物,非键相互作用主导复合物的几何结构。因此,构象生成的理想解决方案应该是同时捕获局部和远距离相互作用。在图 1 中,作者展示了三个典型的分子系统,其中长程相互作用在确定它们的构象方面起着关键作用。
为了实现上述建模远程相互作用的构想,在本文中,作者提出了动态图评分匹配 (DGSM) 用于分子构象生成,遵循CONFGF 的学习原子坐标对数密度梯度的原则。不同于依靠分子图的静态输入作为现有工作,其基本思想是在训练和推理过程中根据原子的空间接近度动态构建原子之间的图结构。这允许模型 (1) 动态学习分子图表示,并考虑到长程相互作用的进化图结构,以及 (2) 动态确定一组对当前原子坐标梯度有贡献的原子间距离。具体来说,动态图的边由两部分组成。第一部分由共价键决定,共价键捕获原子之间的局部相互作用(Ebond、Eangle 和 Etorsion)。边的第二部分由每个训练或采样步骤中原子之间的空间接近度动态确定,即,两个原子只要它们是接近的就连接,无论它们是否键合。这种策略能够有效地捕获非局部相互作用(Enon-bonded),因为长程相互作用的大小与原子之间的距离成反比。同时它保持可扩展性,因为避免连接所有原子对,这具有平方复杂度。此外,建模非键相互作用使模型能够对多分子复合物的构象进行采样,这代表了更广泛的问题适用情况。
作者进行了大量的实验,并将 DGSM 与之前在标准构象生成和属性预测任务上的最先进方法进行了比较。结果表明 DGSM 明显优于以前的方法,证实了建模长程相互作用的好处。此外,为了进一步证明 DGSM 的优势,作者将注意力放在两个更具挑战性的任务上——蛋白质侧链构象预测和多分子复合结构预测。这两项新任务代表了两类实际挑战:预测大分子和多分子复合物的结构。
2
相关工作
先前关于构象生成的工作主要依赖于分子动力学 (MD) ,其中新的构象是根据初始构象和原子间势的物理模型顺序生成的。虽然能够准确地采样平衡构象,但这些方法的计算量很大,尤其是对于大分子系统,例如蛋白质。另一类方法利用距离几何并启发式地将原子之间的距离固定为理想化值,这种方法更快但准确度较低。
最近,人们提出了许多各种用于分子构象生成的深度生成模型,它们在计算效率和准确性之间取得了良好的平衡。在这些方法中,Mansimov 等人首先提出了一个变分自编码器来直接生成 3D 原子坐标。尽管很简单,但这种方法无法模拟分子构象的旋转-平移同变性,导致性能不理想。为了保持旋转-平移同变性,Simm 和 Hernandez-Lobato以及 Xu 等人,首先对分子距离几何进行建模,然后通过解决优化问题从生成的距离重建原子坐标。最先进的方法 CONFGF 估计作用在原子上的伪力并通过 Langevin MCMC 生成构象,绕过了之前工作中的两段式方法,显著提升了性能。这里存在两个同期工作,它们分别通过几何元素组装和双层编程以端到端的方式生成构象。最近也有人尝试使用强化学习进行构象搜索,这种方法无法明确地对键长进行建模,并且与其他方法有着根本的不同。总而言之,之前的所有方法都主要侧重于基于静态输入分子图(或通过在两跳和三跳之外的原子之间添加辅助边来增强图)对局部相互作用进行建模,而忽略了原子之间的长程非键相互作用。相比之下, DGSM 通过动态图评分匹配显式地对局部和长程相互作用进行建模,并有效地解决了上述问题。
3
准备知识
3.1 符号和问题表述

3.2 基于分数的建模生成

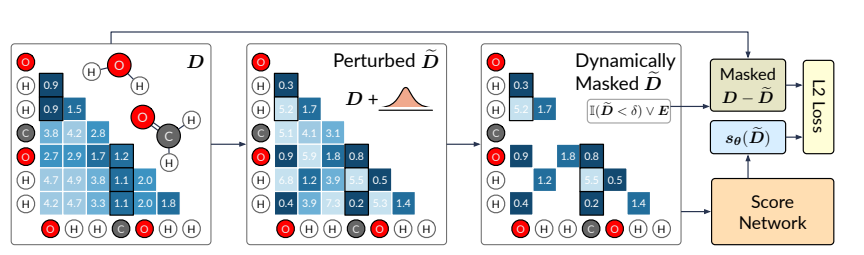
图2 DGSM 的拟训练程序
4
模型
作者的方法将构象生成视为由伪力(即原子梯度)引导的原子依次向高密度区域移动。在Shi等人的带领下,研究者们利用去噪评分匹配来近似原子坐标的对数密度的梯度。为了模拟对局部和长程相互作用(等式 1)敏感的原子梯度,并受到长程相互作用随着距离增加而迅速减少这一事实的启发,作者建议根据当前的空间接近度动态构建在一定距离内的原子对之间具有非键合边的图结构。通过这种方式,使模型能够有效地捕获长程非键相互作用,同时避免连接所有原子,这在计算上是昂贵的。为了确保训练期间图结构的分布与生成期间的分布匹配,作者设计了一种动态图评分匹配算法,其中图结构也在训练期间根据添加的扰动动态确定。整个框架如图2和图3所示。下面作者将在4.1节描述笛卡尔坐标的得分估计框架,在4.2节描述动态图得分匹配算法,在4.3节描述生成过程。
4.1 笛卡尔坐标系下的分数估计
作者的目标是学习原子坐标的对数密度(分数)的梯度,即∇R log p(R | )。使用图神经网络 (GNN) 在绝对笛卡尔坐标上直接参数化评分网络依赖于旋转和平移的选择,这是分子系统中影响构象变化的非必要自由度。因此作者将它们从模型中排除,并首先估计一组动态确定的原子间距离的分数,然后通过到笛卡尔坐标距离的微分来反向传播梯度。

受上述等式的启发,作者首先训练一个噪声条件评分网络来联合预测原子间距离的分数。在训练噪声条件评分网络后,原子坐标的对数密度的梯度 ,可以通过等式4估计。作者有如下命题:

4.2 结合噪声条件评分网络的动态图评分匹配
在本节中,作者描述了所提出的原子间距离动态图评分匹配,目的是对局部和远程相互作用进行建模。为了确保学习的评分函数覆盖具有不同图结构的所有区域,在训练期间基于添加的扰动动态构建具有原子之间非键合边的图结构。继 Songand Ermon之后,作者训练了一个噪声条件评分网络来联合估计一组动态确定的原子间距离的扰动分布的分数,并结合消息传递神经网络(MPNN)将分数参数化。
动态评分匹配 为了捕捉分子系统中非键原子之间的长程相互作用,一种简单的方法是将分子图视为全连接图,并模拟所有原子对之间距离的对数密度梯度。然而,这种做法在计算上是昂贵的,特别是对于大型系统,例如蛋白质,而且有时是不必要的,例如,范德华相互作用会随着距离的增加而迅速衰减。作为补救措施,作者设置了一个截止距离,并假设每个原子只与截止距离内的所有原子相互作用,忽略所考虑球体之外的所有相互作用。这是计算化学中一种非常流行的策略,它在效率和准确性之间取得了良好的平衡。

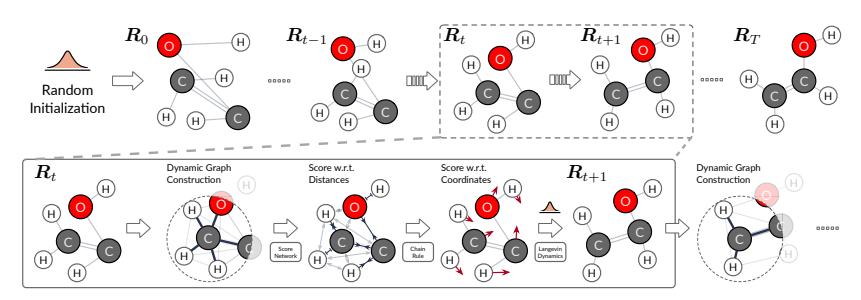
图3 根据朗之万动力学提出的 DGSM 的生成过程
使用上述策略,特定分子图的图结构是可变的并取决于添加的扰动,只要采样足够的噪声,所有图结构都是可能的。这将产生 (1) 一个用于消息传递和表示学习的动态确定的图结构,它考虑了远程相互作用;以及 (2) 一组动态确定的原子间距离,根据等式4它有助于原子坐标的梯度分数估计。请注意等式5 的原始实现需要计算原子对之间的所有距离。在实践中,为了避免二次复杂性,作者在为每个原子添加扰动之前,通过构建具有 2δ阈值的半径图对远邻进行了预过滤,并凭经验验证了这种做法的高效性和有效性。

4.3 生成

然后使用来自评分网络的梯度信息更新构象(方程 4)。这里提供算法1中的伪代码。

5
实验
继先前关于构象生成的工作之后,作者使用以下两个标准任务评估所提出的 DGSM:构象生成(第 5.1 节)和属性预测(第 5.2 节)。为了进一步证明 DGSM 模拟远程相互作用的能力,在两个更具挑战性的基准任务上对其进行了评估:蛋白质侧链构象生成 和 多分子复合物构象生成(第 5.3 节)。在特定任务的部分中描述了实验设置。
5.1 构象生成
设置 此任务通过测量生成的构象的准确性和多样性来评估模型生成稳定分子构象的能力。根据之前的工作,我们使用 GEOM-QM9 和 GEOM-Drugs数据集来完成这项任务。我们使用提供的 train-testsplit。GEOM-QM9 和 GEOM-Drugs 的训练分裂都包含 40,000 个分子,每个分子有 5 个用于训练的构象,或总共 200,000 个构象。GEOM-QM9 的测试分裂包含 200 个具有 22,408 个构象的分子,GEOM-Drugs 的测试分裂包含 200 个具有 14,324 个构象的分子。
将 DGSM 与 5 个最先进的方法进行比较:RDKIT、CVGAE 、GRAPHDG 、CGCF 和 CONFGF。对于测试集中的每个分子,采样的构象是其参考构象的两倍。使用匹配分数 (MAT) 来衡量生成构象的准确性,并使用覆盖分数 (COV) 来衡量多样性。这两个指标都基于分子之间的均方根偏差 (RMSD),同时考虑了对称性。
结果 作者报告了测试拆分 GEOM-QM9 和 GEOM-Drugs 数据集中所有分子的平均 COV 和 MAT 分数。如表 1 所示,DGSM 始终优于其余所有方法。值得注意的是,DGSM 和 CONFGF 都是基于分数的模型,但 DGSM 实现了更好的性能。它们之间的区别在于 DGSM 通过动态图评分匹配成功地考虑了远程相互作用。这证实了建模远程相互作用的显著好处。在图 4 中展示了由不同方法生成的几种构象,这表明 DGSM 成功捕获了高亮区域的远程相互作用,而其他方法失败,导致这些区域的结构扭曲。
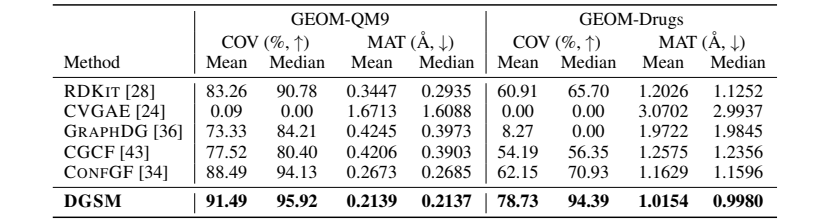
表 1 GEOM-QM9 和 GEOM-Drugs 数据集上的 COV 和 MAT 分数
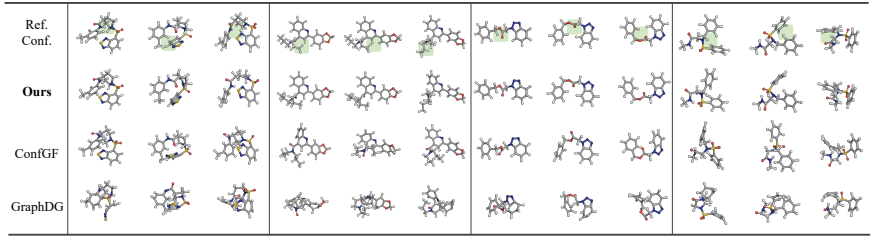
图4 基于来自 GEOM-Drugs 测试集的四个随机分子图,由不同模型生成的构象示例
5.2 属性预测
设置 此任务演示了如何将分子构象的生成模型应用于作为下游任务的特性预测。它还提供了对不同光线下生成的构象质量的评估。通过在之后聚合其构象特性来估计分子图的整体特性。具体来说,首先使用模型为 GEOM-QM9子集中的每个分子图生成 50 个构象,并使用量子化学工具包 PSI4 来计算每个构象的能量和 HOMO-LUMOgap。然后,根据构象能量和间隙计算平均能量 、最低能量 Emin、平均间隙 、最小间隙 和最大间隙 。通过测量它们对实际情况真值的平均绝对误差 (MAE) 来评估估计的集成属性的准确性。CVGAE 被排除在这项任务中,因为它的性能很差。
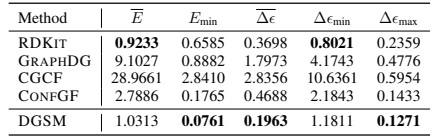
表2 eV 中预测的整体属性的平均绝对误差 (MAE)
结果 表 2 显示 DGSM 明显优于其他基于机器学习的方法。DGSM 对平均能量 和最小间隙 的估计接近 RDKIT,但仍优于最具竞争力的基于 ML 的方法 CONFGF。构象能的计算对几何形状的变化高度敏感——即使键长的细微偏差也会导致显着的能量变化。因此,DGSM 的优越性能表明它比其他方法生成更准确的构象,从而导致更准确的属性估计。这再次验证了建模远程相互作用的有效性。
5.3 大分子和多分子建模
蛋白质侧链构象 此任务是根据其骨架结构预测蛋白质侧链构象。与前几节中的常规分子构象生成相比,这项任务的主要挑战有两个:(1)大量原子,这禁止构建以二次方增长的完整图来模拟长程相互作用。(2)共价键稀疏,这限制了之前工作中边缘增强技术的力量。DGSM 通过引入的动态图评分匹配解决了这两个挑战。
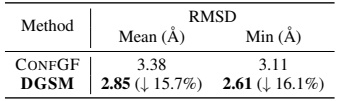
表3 不同的侧链构象生成方法的 RMSD
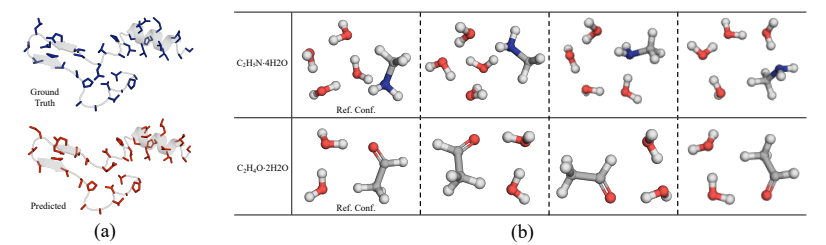
图5 (a) 生成的具有原子级坐标的蛋白质侧链构象的示例 (b) DGSM 生成的两个多分子复合物的构象。
作者使用 SidechainNet 数据集来完成这项任务,并遵循官方的训练测试分割。作者将 DGSM 与最先进的构象生成模型 CONFGF 进行了比较。此任务的主要目的是证明 DGSM 对大分子的有效性。对于每个蛋白质,生成了 5 个不同初始化的侧链构象,并计算了真实构象和生成的构象之间的均值和最小 RMSD。通过对表 3 测试集中每种蛋白质的分数求平均值来报告总体平均和最小 RMSD 分数,表明了 DGSM 比以前的最先进模型取得了更好的性能。作者还在图5(a)中给出了一个例子,可以看到预测的构象在主要部分与实际情况是一致的。
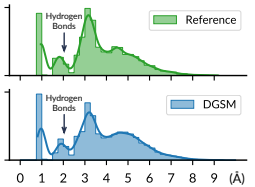
图6 氢氧距离的分布
多分子复合物构象 此任务是预测多分子复合物的构象。多分子复合物由多个分子组成,它们之间没有共价键。长程相互作用支配着多分子复合物的结构。此任务的目的是展示 DGSM 对更广泛问题的潜在应用,并为构象生成提供新的基准。作者使用量子化学软件 xtb 构建了一个由24个水-有机复合物组成的数据集,每个水-有机复合物都有数百个构象,其中20个复合物用于训练模型,剩下4个作为测试数据集。不报告基于 RMSD 的指标,例如 COV 和 MAT,因为多分子复合物的结构非常灵活。图 5(b) 显示了两组生成示例。作者观察到水分子规则地放置在溶质有机分子周围。值得注意的是,氢键(水和溶质之间,以及水和水之间)正确形成了。这也可以在氢氧距离的直方图(图 6)中得到证明,其中在 1.5Å 和 2.5Å 之间有一个峰值,即氢和氧之间的氢键长度范围。
6
结论和未来工作
作者提出了 DGSM,这是一种用于生成平衡态分子构象的基于评分的新方法。通过在训练和推理期间基于原子之间的空间接近度动态构建图结构,DGSM 能够对分子系统中的局部和远程相互作用进行建模。作者还设计了一种动态图评分匹配算法来有效地估计原子梯度,其中图结构是根据添加的扰动动态确定的。对两个标准任务和两个原始任务的大量实验表明,DGSM 大大优于最先进的方法,证实了建模远程相互作用的显著优势。将来,作者计划将该方法应用于更具挑战性的蛋白质结构预测问题。
参考资料
Shitong Luo, Chence Shi, Minkai Xu, Jian Tang. Predicting Molecular Conformation via Dynamic Graph Score Matching。