原位电子显微学探索固体中的离子迁移行为(二)
作者:陈树林1,2 高鹏1,3,4,5
(1 北京大学物理学院电子显微镜实验室)
(2 哈尔滨工业大学先进焊接与连接国家重点实验室)
(3 北京大学物理学院量子材料科学中心)
(4 量子物质科学协同创新中心)
(5 固态量子器件北京市重点实验室)

摘要 离子在固体材料中的迁移是很多应用的基础,包括锂离子电池、存取器件及催化剂等。研究固体材料中的离子迁移行为是固态离子学的核心内容。微观上,固体中离子迁移的行为取决于材料微观结构所确定的局域势垒特征。因此,研究离子迁移行为与微观结构的关联至为重要。文章讲述了具有高空间分辨率的原位透射电子显微镜技术用于锂离子电池材料、阻变存储材料等体系中离子迁移行为的研究现状与发展趋势。
关键词 原位透射电镜,离子迁移,锂离子电池,阻变存储器,催化剂,太阳能电池
3.1 存储器
存储器件是信息科学里最重要的单元之一。尤其在大数据时代的今天,我们对存取器件的要求也是越来越高,一个好的存储器需要有几个特点:容量大、制作成本低、功耗低、稳定性高。其中,阻变存储器,即“电阻转变式随机存储器”(Resistive Random Access Memory,RRAM),具有存储密度高、结构简单、读写时间短以及3D存储潜力等优点,引起了半导体行业的广泛关注[34,35]。阻变存储器主要利用某些薄膜材料在电激励的作用下会出现高、低阻态的特征,并且这些电阻态能够转变的现象来进行数据的存储。RRAM器件一般具有“金属—绝缘体—金属”的三明治结构,这种三明治结构的绝缘层可以是二元或多元的金属氧化物、硫属化合物和有机化合物等。
根据阻变存储器的阻变机制,阻变存储器可以分为阴离子型阻变存储器和阳离子型阻变存储器。对于阻变机制的解释有很多模型,最被人广泛接受的是导电通道型阻变机制[36],该机制认为器件的高低阻态由介质薄膜中导电通道的连接与断开而决定。比如阳离子型阻变存储器,其初始态一般是高阻态,经过一个Forming 过程才能进入高低阻态转换的工作状态。在活性金属电极一端加上一个足够大的电压,金属失去电子,变成金属阳离子进入电解质,并在电场作用下迁移至阴极,获得电子被还原成金属原子。随着金属原子在电解质中的不断沉积,最终导致这些金属原子形成一个或多个金属通道贯穿整个电解质,连接顶底两电极,此时器件变成低阻态,定义为ON,如图5(a)所示。在低阻态器件上加一个反向的电压(双极型器件)或同向的更大的电流(单极型器件),使导电通道由于电化学迁移或者热效应而断开,器件便转变回高阻态,定义为OFF,如图5(b)所示。通过阻变存储器的高低阻态的保持,可以实现二进制数据的存储。这类基于阳离子氧化还原的阻变存储器一般又被称作电化学金属化存储器(Electrochemical Metallization Memory,ECM),或导电桥连随机读取存储器(Conductive Bridge Random Access Memory,CBRAM)。

图5 阻变存取器件中的导电细丝模型[38] (a)开的状态(“1”)对应于短路,导电细丝将两个电极连接起来;(b)关的状态(“0”)对应于开路,导电细丝断开;(c)Forming 过程后对应的TEM图;(d)阻变存取器中的导电细丝模型;(e)原位观察导电细丝形成过程的TEM图
虽然在实验中已经多次观察到这种导电通道,但是其微观图像及动力学过程仍然不清晰。关于导电通道型阻变机制,大家关心的核心问题是:(1)导电通道的化学成分和微观结构,不同材料体系中导电通道的化学成分和微观结构均存在差异;(2)导电通道的生长、断裂的动力学过程,这个动力学过程决定了读写速率、数据存取寿命等。同样,在不同的体系中,导电通道的动力学演化行为是不同的。有很多研究者试图用不同的表征手段来研究这些内容,比如电学测量、导电原子力显微镜(LC-AFM)、红外热成像、X射线微区荧光和X射线近边吸收谱等。考虑到导电通道的尺寸在纳米量级,在这些研究手段中,只有LC-AFM具有足够高的空间分辨率,其他手段都是宏观测量,得到的仅是宏观信息的平均结果。而LC-AFM是一种表面探测技术,只能表征表面的状态,用来证明导电通道存在与否,却无法深入研究导电通道本身的性质。原位透射电子显微学方法则可以弥补LC-AFM的不足,真正在原子尺度对导电通道的微观结构与化学成分进行表征[37]。
图5(c)所示为阻变存储器经过Forming 过程后对应的显微结构[38]。在电场作用下,活性Ag电极产生的Ag+逐渐沉积形成Ag金属导电细丝,使得两电极直接相连,电流瞬间降低。这些导电细丝一般只有一条连通正负级,其余的导电细丝尚未连接起正负极。这是由于细丝生长过程是一个自我限制的过程,一旦有一根细丝连通正负极,其余细丝的生长便会因为电路导通后电场强度的降低而受到限制。由于靠近Pt 电极处的电势更低,导电细丝倾向于从Pt 端开始逐渐向Ag电极端生长。此外有两点与传统ECM理论[39]不符合的现象:一是,导电细丝是由离散的颗粒组成,而ECM理论则认为导电通道是连续的形貌;二是,最细的导电细丝靠近Pt 电极端,而ECM理论则认为导电细丝一般在靠近活性电极Ag 的那一侧更细。细丝的几何形状对EMC实际打开与关断的写入操作尤为重要,这是因为金属细丝的溶解以及形成总是在最细的部位进行的。通过观察器件断开(OFF)后的显微结构,可以发现在电介质和Pt 的界面处的导电细丝会先溶解[38]。通常这些导电细丝不会完全溶解,这是由于当电介质和Pt 界面处最细的细丝断裂后,电路断开,引起氧化还原反应的效率降低。在器件断开(OFF)后再次打开(ON)时,电介质中残留的细丝便会促进后续的细丝生长。通过构建W/a-Si/Ag“三明治”结构,用原位TEM观察了导电细丝的生长过程,如图5(e)所示。金属钨针尖上包覆的非晶硅使得导电细丝必须穿过a-Si 层后才可以实现Ag 电极和W针尖的电导通。因而可以观察到导电细丝的生长是始于活性Ag 电极一侧而逐渐向惰性W电极一侧生长。这些Ag 颗粒也呈现圆锥型,并且朝Ag 电极那一侧的要宽一些。这些实验结果说明导电细丝的生长受电解质类型的影响,这是由于金属离子在不同电解质中的迁移势垒不同造成的。
这些现象一方面解释了在阻变存储器中一般首次Forming 所需的电压阈值要高于后续的ON/OFF过程,另一方面表明了在实际工作中,器件的电介质和嵌入电极(Pt)的界面比电介质和活性电极(Ag)的界面可能更加重要。这些有助于加强我们理解基于导电细丝模型的阻变机制,并指导后续ECM材料和器件的优化。
3.2 催化剂
汽车尾气污染是导致雾霾和全球变暖的因素之一,通常采用催化剂对汽车尾气进行净化。二氧化铈(CeO2)因其在三效催化剂(TWC)方面上的应用而备受关注[40]。CeO2在TWC 中的核心工作原理是铈能够以正三价(+3)和正四价(+4)的形式存在,能够在较高温度下发生氧化/还原反应。然而二氧化铈的工作温度高,使得在冷启动条件下,汽车的尾气无法完全被净化,因此很多研究致力于降低氧化铈的工作温度,进而提高催化剂在低温下的性能,减少冷启动条件下的尾气污染[41]。通过加入氧化锆和稀土氧化物,可以有效提高二氧化铈的低温氧化还原活性和稳定性;此外也可以通过氧空位的构建来提高二氧化铈的低温催化性能,通常的做法是将其加热至高温处理,使之释放出氧气,形成氧空位,从而提高二氧化铈的催化性能。CeO2是一种离子键化合物,其阳离子晶格非常稳定,而阴离子相对活泼,容易发生迁移[42]。考虑到氧离子带负电荷,在电场下会受到电场力的作用,而同时氧离子又有很高的扩散系数,因此有可能通过电场力产生氧空位并驱动其迁移,这样就可以降低CeO2发生氧化还原反应的温度,从而解决汽车冷启动的尾气污染问题。
图6 显示了原位TEM研究室温电场驱动CeO2中的可逆氧化还原反应过程,记录了在电场周期性作用下二氧化铈在原子尺度上的结构演变[43]。图6(a)是在铌掺杂钛酸锶上外延生长的CeO2薄膜的高分辨TEM图片。当加上一个6 V的偏压后,会有“水纹”状的条纹出现,如图6(b)所示。这些条纹从阴极附近产生,然后穿过整个薄膜,最后到达阳极。通过电子能量损失谱和电子衍射数据分析,可以证明带有“水纹”状的条纹的样品其实是Ce2O3。即在电场作用下,氧化铈中的氧离子由于扩散势垒降低而向阳极迁移,在阳极表面变成氧气逃脱从而产生氧空位,使得CeO2中有四分之一的氧离子缺失,氧空位带正电,在电场作用下聚集到阴极,形成“水纹状”的Ce2O3。从CeO2和Ce2O3两者的能带结构也可以发现CeO2中的4f 轨道是空的,转变成Ce2O3后,4f 轨道填充了部分电子,使得禁带宽度从6 eV[44]降低到2.4 eV[45],如图6(c,d)所示,从而提高铈氧化物的导电性能。
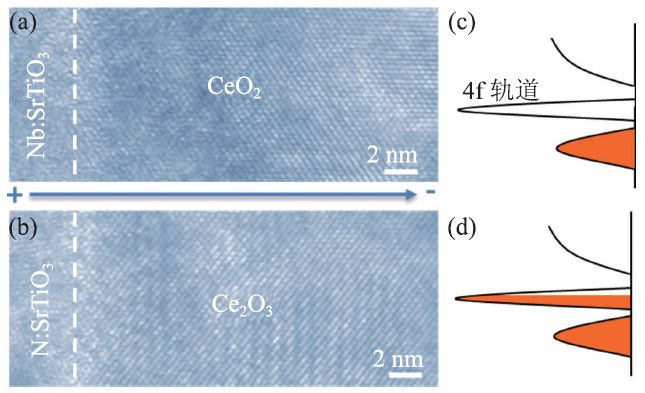
图6 CeO2中氧离子在电场作用下的迁移[43] (a,b)CeO2与Ce2O3高分辨TEM 图片;(c,d)CeO2与Ce2O3对应的能带结构示意图。CeO2中的氧离子在电场作用下迁移,引起CeO2到Ce2O3的结构转变
原位TEM观察结果表明,在电场的驱动下,大量的氧原子可以被释放和储存在氧化铈中[46,47]。这些结果对CeO2基催化剂的应用有重要的启示,即电场可以降低催化剂的工作温度,从而克服内燃机冷起动的问题。除了催化作用外,氧化铈的电驱动氧化还原反应为此类离子导体在中低温固态燃料电池中的应用开辟了一条新的途径。
研究扩散机制是材料科学中的一个经典问题。但是直接观察扩散过程是比较困难的,多数实验探索仅限于对浓度的宏观测量。最近,借助于球差矫正透射电镜,我们可以直接观察体相中的离子扩散[48]。其机理是,透射电镜中的高能电子可与样品中的原子发生碰撞,转移给原子的能量可以驱动其从一个平衡位置迁移到另一个平衡位置,进而引发相变,甚至结构分解[49]。从这个意义上说,电子束辐照与光学辐照、加热、电场之间具有较大的相似性,在这些过程中离子都是从外场获得能量或者离子的势垒变低,从而克服迁移势垒而触发相变。本章节将简单介绍在电子束辐照下钙钛矿太阳能电池材料和锂离子电池材料中的离子扩散和相变过程。
4.1 电子束辐照下CH3NH3PbI3中的离子迁移
近年来,由于合成价格低廉、光电转化效率高,基于有机无机杂化钙钛矿材料(CH3NH3PbI3,MAPbI3)的太阳能电池得到了飞速发展[50—52]。其光电转化效率在近十年内已从最初的3.8%迅速增长至目前的23.7%。据报道,钙钛矿在高温[53]、氧气[54]、潮湿[55]和光照条件[56]下会迅速降解,这严重阻碍了该技术的产业化。这些结构的不稳定性与材料内部的离子迁移情况息息相关。研究者发现,在加热条件下,有机无机杂化钙钛矿材料中的碘离子和铅离子会相继发生扩散,导致最终分解成碘化铅[53]。此外在电场和光照条件下的离子迁移也会引起J—V曲线的迟滞。一般认为,这些器件在实际工作过程中由外场驱动的离子迁移导致的结构退化是影响器件性能下降和寿命缩短的直接原因。因此,研究钙钛矿材料中离子迁移导致的结构不稳定性和相变过程具有重要意义。
由于钙钛矿材料对电子束十分敏感,其原子成像充满挑战[57]。因此,研究者们多采用电子束剂量相对较弱的电子衍射模式用于判定钙钛矿的结晶性以及相成分。图7所示为通过连续电子衍射图谱的变化研究了MAPbI3 在电子束辐照下的分解路径[58]。当电子束剂量率控制在1 e Å-2 s-1时,首先可以采集到原始的钙钛矿的电子衍射图(图7(a)),当剂量逐渐增加到328 e Å-2时,电子衍射图中含有大量超结构衍射点,对应着超结构相的生成(图7(b)),最终当剂量增加到2191 e Å-2,结构完全分解成碘化铅(图7(c))。考虑到碘离子在MAPbI3中扩散势垒较低,容易发生迁移,根据电子衍射,可推断超结构相的产生很可能是由有序碘空位的形成引起的,如图7(d,e)所示。
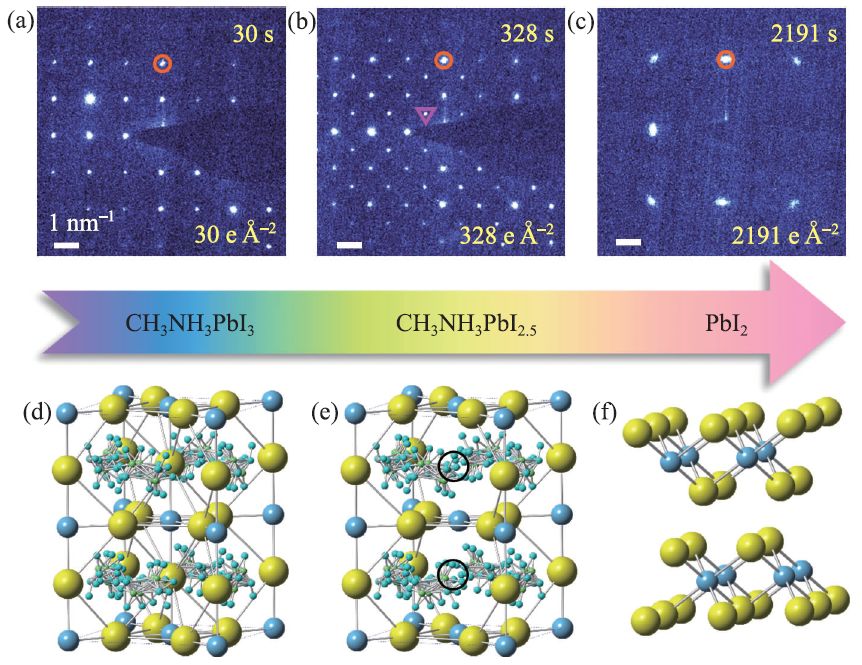
图7 电子束辐照下CH3NH3PbI3中碘离子的迁移[58] (a—c)在1 e Å-2 s-1的电子束辐照剂量率下,CH3NH3PbI3的选区电子衍射图的演变过程;(d—f)依次为CH3NH3PbI3,超结构相CH3NH3PbI2.5,PbI2的原子结构示意图,黄色球代表I,蓝色球代表Pb,原子簇代表CH3NH3,黑色圆圈代表有序的碘空位
此外,形成具有超结构中间相MAPbI2.5可以使得碘离子之间的排斥作用和弹性应变能最小化,这与锂离子电池材料中的“阶(staging)”现象类似[22]。增大电子束辐照的剂量,超结构相MAPbI2.5 会进一步分解为PbI2。这种直接观测有助于我们更好地理解钙钛矿材料在实际工作过程中由离子扩散导致的结构相变。此外,可以发现MAPbI3和PbI2的衍射图谱较为接近,碘化铅的(01-4)衍射点和MAPbI3的(22-0)衍射点在倒空间中距离十分接近,所以当忽略了MAPbI3中消失的(11-0)和(110)衍射点时,就容易把碘化铅误标定成MAPbI3。以往的许多研究就忽略了这些细微的变化,错误地将分解后的PbI2识别为MAPbI3[59—62]。因此,在分析这些电子束敏感材料的电子显微学数据时必须特别注意。
4.2 电子束辐照下锂离子电池材料中的离子迁移
对于可充放电的锂离子电池而言,当进行充放电的时候,希望电极材料在反复脱锂和嵌锂过程中,其结构框架保持不变。然而在实际的脱嵌过程中,电极材料中的某些金属离子会变得活泼进而移动引起结构相变。这些结构的改变有可能阻塞锂离子扩散通道,造成电池容量衰减以及电压滞后[63]。由于这种过渡金属离子扩散也可以在电子束作用下激发,所以有研究者专注于电子束辐照下的相变研究。比如正极材料LiMn2O4在电子束辐照下,会发生尖晶石至盐岩结构的相变[64]。利用球差矫正透射电镜高的空间分辨率这一优势,可以观察到尖晶石至盐岩结构的相变过程涉及锂离子、锰离子以及氧离子的迁移。具体为:处在四面体中心的锂离子会向相邻共面的八面体中心的空位处迁移,处在八面体中心的锰离子会先迁移到氧四面体中心位置作为中间过程,然后再迁移到相邻共棱的八面体中心。在这个过程中,氧离子则会向对称性更高的位置略微偏移,保持整个阴离子结构框架基本不变。
通过在原子尺度观测离子扩散的动态过程和化学环境的演变,有助于我们更好地理解锂离子电池材料的性能衰减问题,并可以指导我们通过元素掺杂以及表面修饰去稳定结构并优化性能。为研究扩散机制提供了一种新的方法,不仅限于表面现象,而且适用于体材料内部的扩散机制。
固体中的离子迁移是决定材料结构、加工和性能的基本过程之一,在决定器件寿命方面也起着至关重要的作用。通过原位TEM技术可以实时地观察到锂离子电池、阻变存储器、催化剂、太阳能电池等器件中的材料内部发生的离子迁移行为以及相变过程,有助于我们探究微观上的离子扩散行为与宏观上的器件性能之间的相互联系,更好地理解器件的工作原理及其失效机制,从而为优化器件性能提供理论指导。考虑到实际的器件工作的环境往往同时存在多个外场,因此将来通过对原位电镜技术改进,实现在多场耦合条件下探测局域固态离子迁移,具有重大意义。此外,目前原位局域场探测技术的稳定性还有待提升,将来更高的稳定性可以实现外场条件下原子分辨的表征,有助于解析更精细的结构相变。在电镜实验中,总是无法避免电子束辐照的影响,将来使用更好的相机或者探测器在低剂量下成像将有效减小电子束辐照的影响,具有更高时间分辨的相机或者探测器也能够使得我们得到原位动力学过程更多的细节。已经报道的大部分原位电镜实验结果都是关于相变结构的转变,实际上电镜也具有强大的电子结构表征功能,现在基于冷场单色仪球差校正电镜能够在原子尺度上实现亚10 meV的电子能量损失谱学功能[65],可以在表征离子迁移的同时探测其对应的电子结构变化。
致谢 感谢南方科技大学俞大鹏老师和中国科学院物理研究所白雪冬老师的大力支持及有益讨论,以及丁正平博士和屈可博士给出的宝贵意见。
参考文献
[1] Paul A,Laurila T,Vuorinen V et al. Thermodynamics,diffusion and the Kirkendall effect in solids. Cham:Springer International Publishing,2014
[2] Park M,Zhang X,Chung M et al. J. Power Sources.,2010,195:7904
[3] Mehrer H. Diffusion in solids:fundamentals,methods,materials,diffusion-controlled processes. New York:Springer Science &Business Media,2007
[4] Kühne M,Börrnert F,Fecher S et al. Nature,2018,564:234
[5] Huang J Y,Zhong L,Wang C M et al. Science,2010,330:1515
[6]Wang F,Yu H,Chen M et al. Nat. Commun.,2012,3:1201
[7] He K,Zhang S,Li J et al. Nat. Commun.,2016,7:11441
[8] Li Y,Yan K,Lee H et al. Nat. Energy,2016,1:15029
[9] Yan K,Lu Z,Lee H et al. Nat. Energy,2016,1:16010
[10] Yuan Y,Nie A,Odegard G M et al. Nano Lett.,2015,15:2998
[11] He K,Xin H L,Zhao K et al. Nano Lett.,2015,15:1437
[12] Liu X H,Wang JW,Huang S et al. Nat. Nanotech.,2012,7:749
[13] Zhu Y,Wang JW,Liu Y et al. Adv. Mater.,2013,25:5461
[14] Chhowalla M,Shin H S,Eda G et al. Nat. Chem.,2013,5:263
[15] Chen R,Luo R,Huang Y et al. Adv. Sci.,2016,3:1600051
[16] Seo JW,Jang J T,Park SWet al. Adv. Mater.,2008,20:4269
[17] Gao P,Wang L,Zhang Y et al. Nano Lett.,2016,16:5582
[18] Safran S A,Hamann D R. Phys. Rev. Lett.,1979,42:1410
[19] Dahn J R,Fong R,Spoon M J. Phys. Rev. B,1990,42:6424
[20] Safran S A,Hamann D R. Phys. Rev. B,1980,22:606
[21] Gu L,Zhu C,Li H et al. J. Am. Chem. Soc.,2011,133:4661
[22] Fernandez-Moran H,Ohstuki M,Hibino A et al. Science,1971,174:498
[23] Momma T,Shiraishi N,Yoshizawa A et al. J. Power Sources,2001,97-98:198
[24] Gao P,Zhang Y,Wang L et al. Nano Energy,2017,32:302
[25] Chen S,Wang L,Shao R et al. Nano Energy,2018,48:560
[26] Li Y,Wu D,Zhou Z et al. J. Phys. Chem. Lett.,2012,3:2221
[27] Su J C,Pei Y,Yang Z H et al. RSC Adv.,2014,4:43183
[28] Lu J M,Zheliuk O,Leermakers I et al. Science,2015,350:1353
[29] Yuan H,Bahramy M S,Morimoto K et al. Nat. Phys.,2013,9:563
[30] Silbernagel B G,Thompson A H,Gamble F R. AIP Conf. Proc.,1975,24:380
[31]Waszczak J V,Disalvo F J. Phys. Rev. B,1981,23:457
[32] van Bruggen C F,Haas C. Solid State Commun.,1976,20:251
[33] Shao R,Chen S,Dou Z et al. Nano Lett.,2018,18:6094
[34] Akinaga H,Shima H. P. IEEE,2010,98:2237
[35]Waser R,Aono M. Nat. Mater.,2007,6:833
[36] Kwon D,Kim K M,Jang J H et al. Nat. Nanotech.,2010,5:148
[37] Tian X,Yang S,Zeng M et al. Adv. Mater.,2014,26:3649· 178
[38] Yang Y,Gao P,Gaba S et al. Nat. Commun.,2012,3:732
[39] Waser R,Dittmann R,Staikov G et al. Adv. Mater.,2009,21:2632
[40] Lawrence N J,Brewer J R,Wang L et al. Nano Lett.,2011,11:2666
[41] Shelef M,Mccabe RW. Catal. Today,2000,62:35
[42] Bevan D J M,Kordis J. J. Inorg. Nucl. Chem.,1964,26:1509
[43] Gao P,Kang Z,Fu Wet al. J. Am. Chem. Soc.,2010,132:4197
[44] Wuilloud E,Delley B,Schneider Wet al. Phys. Rev. Lett.,1984,53:202
[45] Prokofiev A V,Shelykh A I,Melekh B T. J. Alloy. Compd.,1996,242:41
[46] Li X,Qi K,Sun M et al. Appl. Phys. Lett.,2015,107:211902
[47] Li X,Qi K,Sun M et al. Chem. Cat. Chem.,2016,8:3326
[48] Ishikawa R,Mishra R,Lupini A R et al. Phys. Rev. Lett.,2014,113:155501
[49] Egerton R F,Li P,Malac M. Micron,2004,35:399
[50] Li X,Bi D,Yi C et al. Science,2016,353:58
[51] Chen W,Wu Y,Yue Y et al. Science,2015,350:944
[52] Burschka J,Pellet N,Moon S et al. Nature,2013,499:316
[53] Divitini G,Cacovich S,Matteocci F et al. Nat. Energy,2016,1:15012
[54] Aristidou N,Eames C,Sanchez-Molina I et al. Nat. Commun.,2017,8:15218
[55] Yang J,Siempelkamp B D,Liu D et al. ACS Nano,2015,9:1955
[56] Bryant D,Aristidou N,Pont S et al. Energy Environ. Sci.,2016,9:1655
[57] Zhang D,Zhu Y,Liu L et al. Science,2018,359:675
[58] Chen S,Zhang X,Zhao J et al. Nat. Commun.,2018,9:4807
[59] Zhu H,Fu Y,Meng F et al. Nat. Mater.,2015,14:636
[60] Son D,Lee J,Choi Y J et al. Nat. Energy,2016,1:16081
[61] Li D,Wang G,Cheng H et al. Nat. Commun.,2016,7:11330
[62] Fan Z,Xiao H,Wang Y et al. Joule,2017,1:548
[63] Mohanty D,Li J,Abraham D P et al. Chem. Mater.,2014,26:6272
[64] Gao P,Ishikawa R,Tochigi E et al. Chem. Mater.,2017,29:1006
[65] Li Y,Wu M,Qi R et al. Chin. Phys. Lett.,2019,36:26801
本文选自《物理》2019年第3期






