Google Research进军蛋白质结构预测:为Pfam数据库新增680万标注数据
![]()
新智元报道
新智元报道
编辑:LRS
【新智元导读】用深度学习模型来预测蛋白质的结构和功能已经取得了不小的进展,但还缺乏优质的数据。最近Google开源了一个模型ProtENN,提供了680万条蛋白质结构数据Pfam-E,约等于之前十年的工作量。
蛋白质是所有生物体中的重要分子,在我们身体的结构和功能中都发挥着核心作用。并且从药物到洗衣粉等日常生活用品中,蛋白质也无处不在。
虽然每个蛋白质都是由氨基酸构成的链,但不同的氨基酸序列导致了不同的蛋白质结构,也导致了不同蛋白质具有不同的功能。
了解蛋白质的结构和功能之间的关系,是一项具有深远科学意义的长期研究。
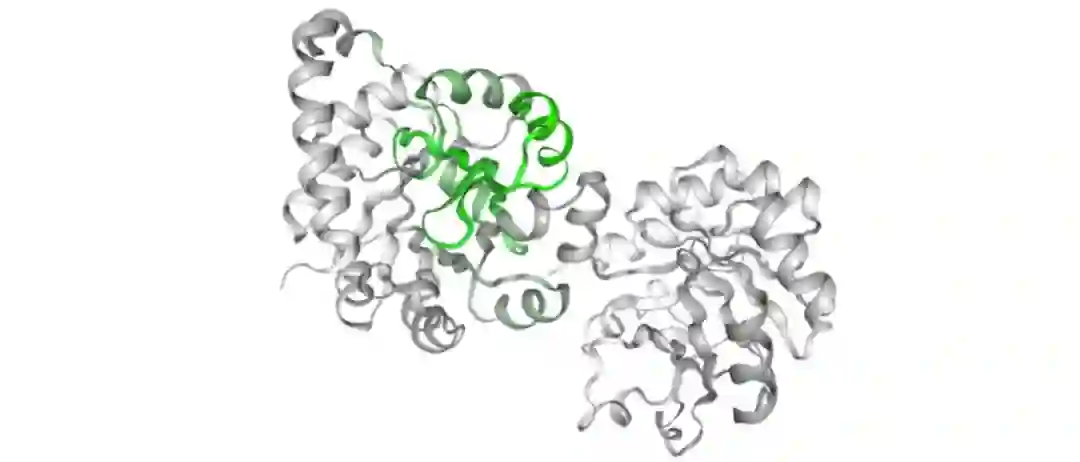
2018年,DeepMind推出第一版AlphaFold模型,采用深度学习+传统算法结合的方式,借助大算力的优势,成功取得第13届蛋白质结构预测CASP竞赛的冠军,AlphaFold仅需数天即可完成科学家数年的工作。
而后2020年的AlphaFold2模型则使用更大的算力,训练更大的模型,准确率远远超越其他竞争对手,也正式掀起了大规模深度学习模型进行蛋白质结构预测的热潮。
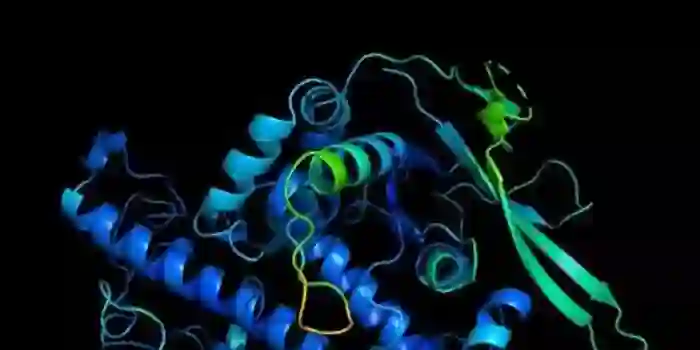
除了广为人知的AlphaFold外,科学界在使用计算工具直接从序列中推断蛋白质功能方面也有很长的历史。
例如,著名的蛋白质家族数据库Pfam包含许多高度详细的计算注释,描述了一个蛋白质域的功能,如球蛋白和胰蛋白酶家族。
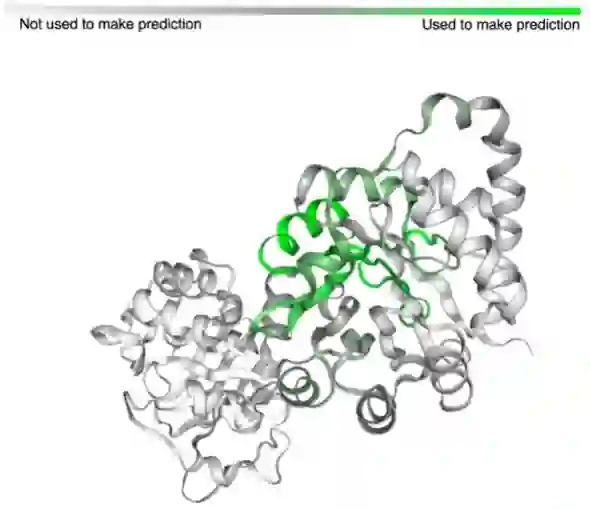
虽然现有的方法已经成功地预测了数以亿计的蛋白质的功能,但仍然有许多功能未知的蛋白质,研究显示,至少有三分之一的微生物蛋白质没有得到可靠的注释。
随着公共数据库中蛋白质序列的数量和多样性继续迅速增加,准确预测高度多样化氨基酸序列的功能变得越来越紧迫。
最近,Google Research在Nature Biotechnology(近两年影响因子54.908)上发表了一篇论文,提出了一个机器学习模型ProtENN,能够可靠地预测蛋白质的功能,并且为Pfam新增了大约680万条蛋白质功能注释,大约相当于过去十年进展的总和。研究人员把新数据集发布为Pfam-N。

为了鼓励在这个方向上的进一步研究,研究人员发布了ProtENN模型和一篇类似distill的交互式文章。用户只需要在该互动工具输入一个序列,就能够在浏览器中实时获得预测的蛋白质功能的结果,而不需要其他设置。

结构预测就是分类
结构预测就是分类
在计算机视觉中,常用的流程就是先为图像分类任务训练一个模型,如CIFAR-100,然后将其作为预训练模型再扩展到更具体的任务,如物体检测和定位。
研究人员也采用这种模式,先开发了一个蛋白质结构域的分类模型,作为下一步对整个蛋白质序列进行分类的模型的预训练。
在训练过程中,把这个问题看作是一个多类分类任务,类别标签包含所有从Pfam数据库中提取的17929个类。
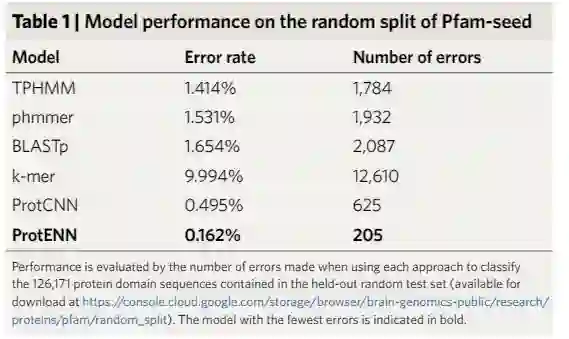
下一步就需要一个模型将蛋白质序列与蛋白质功能联系起来。
虽然目前有许多模型可用于蛋白质结构域分类,但它们都一个明显的缺点:基于线性序列的排列,而没有考虑蛋白序列中不同部分的氨基酸之间的相互作用。蛋白质并不只是停留在一排氨基酸上,它们会自行折叠,这样不相邻的氨基酸就会对彼此产生强烈的影响。
一些sota模型会将新的查询序列(query sequence)与一个或多个具有已知功能的序列进行比对。
但如果新序列与任何具有已知功能的序列高度不相似的话,那这种对具有已知功能的序列的依赖性就会使得预测一个新序列的蛋白质功能更具有挑战性。
此外,基于对齐的方法是计算密集型的,如果想要把这个算法应用于大型数据集,如元基因组数据库MGnify,其中包含超过10亿条蛋白质序列,成本过高的话就失去了实用价值。
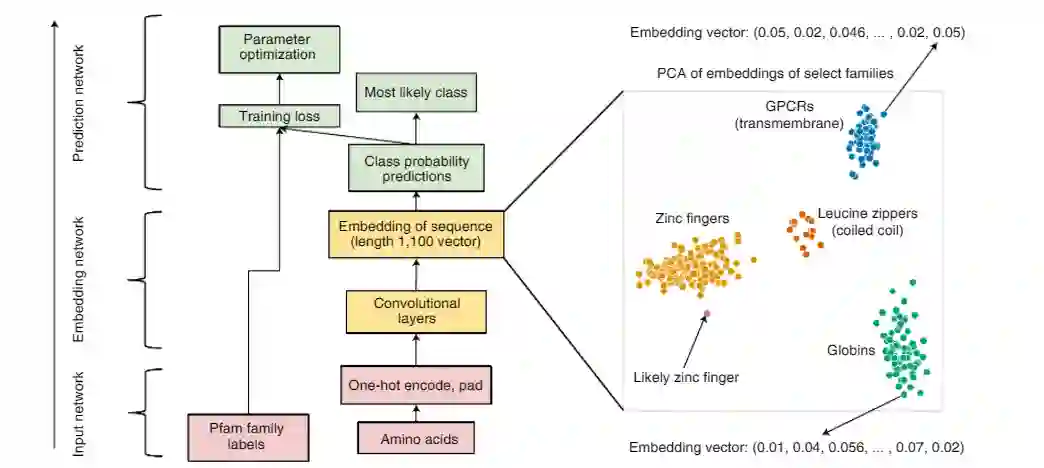
为了应对这些挑战,研究人员建议使用卷积神经网络(CNN),很适合于模拟非局部的成对氨基酸相互作用,并且可以在GPU硬件上快速运行。
研究人员训练一维CNN来预测蛋白质序列的分类,称之为ProtCNN;以及多个独立训练的ProtCNN的集成模型,称之为ProtENN。
这种方法的目的是通过开发一种可靠的ML方法,补充传统的基于对齐的方法,为了证明效果,研究人员还提出了一种方法来测量预测准确性。
蛋白质的进化也要考虑
蛋白质的进化也要考虑
与其他领域的分类问题类似,蛋白质功能预测的挑战不在于为任务开发一个全新的模型,而在于创建公平的、大规模的训练和测试集,以确保模型对未见过的数据做出准确的预测。
由于蛋白质基本都是从共同的祖先演变而来的,不同的蛋白质往往共享其氨基酸序列中的相当大的一部分。如果没有特意调整数据分布,测试集可能会被与训练数据高度相似的样本所支配,这也可能会导致模型通过简单地「记忆」训练数据就能准确预测,而没有学会更广泛地归纳。
为了防止这种情况的出现,必须使用多个独立的设置来评估模型的性能。对于每一次评估,研究人员将模型的准确性作为每个被保留的测试序列与训练集中最近的序列之间的相似性的函数来分层。
第一个评估包括一个聚类的分割训练和测试集,蛋白质序列样本按序列相似度进行聚类,整个聚类被放入训练集或测试集。因此,每个测试实例与每个训练实例至少有75%的差异。这个任务下的更强的性能表明,一个模型可以概括地对分布外的数据做出准确的预测。
在第二个评估中,研究人员使用随机分割的训练和测试集,根据对实例分类难度的估计对其进行分层。难度的衡量标准包括测试例子和最近的训练例子之间的相似性,以及来自真实类别的训练例子的数量(只给少量的训练例子,要准确预测功能就更难了)。
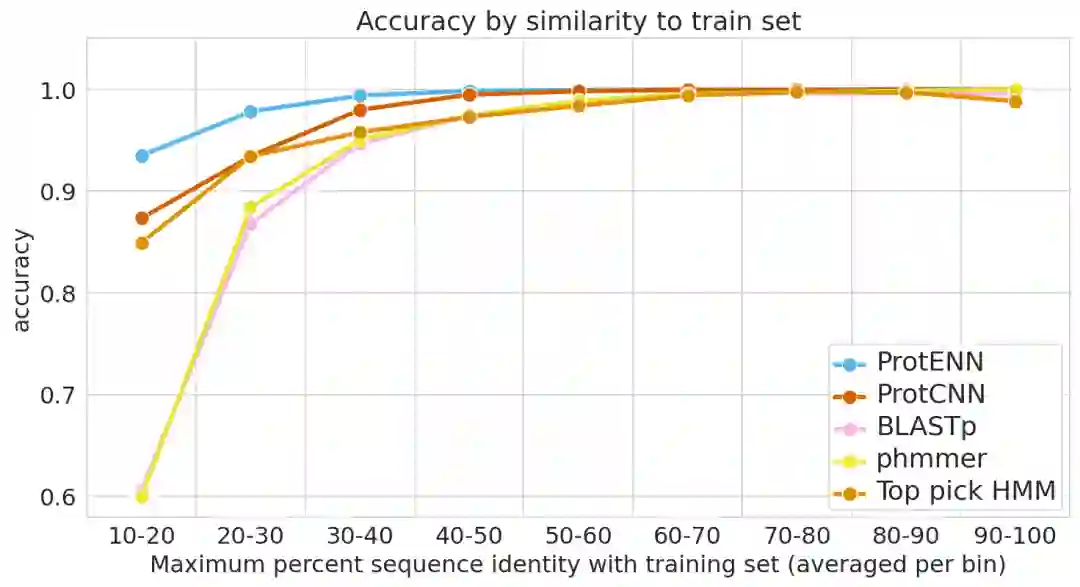
除此之外,研究人员还与Pfam团队合作,测试文中提出的方法学概念证明是否可用于标记真实世界的序列。结果证明了ProtENN可以学习到基于对齐的方法的补充信息,比任何一个方法学到的信息都要多。
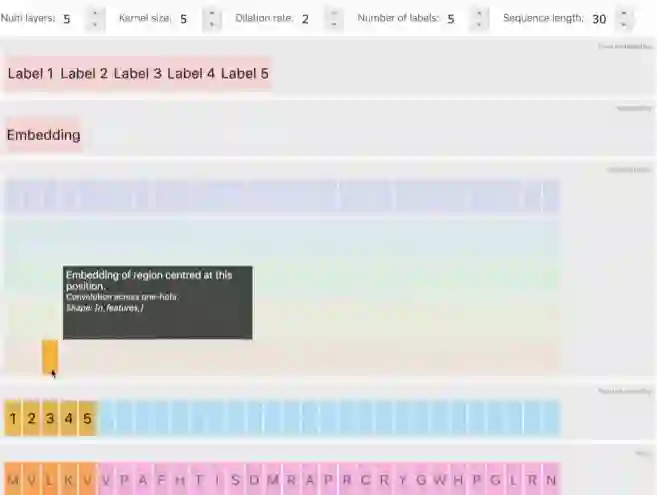
在看到这些方法和分类任务的成功后,研究人员还建立了一个工具,使用户能够探索模型预测、embedding和输入序列之间的关系,在前文提到的交互式网页中可以体验这项功能,可以发现类似的序列在embedding空间中被聚在一起。
从AlphaFold和CAFA到会议上专门讨论这一主题的大量研讨会和研究报告,深度学习预测蛋白质的相关工作也逐渐增加。
研究人员认为在这项工作的基础上,可以继续与整个领域的科学家合作,利用他们的专业知识和数据,结合机器学习模型的进步,将帮助人类进一步揭示蛋白质的世界。
参考资料:
https://ai.googleblog.com/2022/03/using-deep-learning-to-annotate-protein.html
https://google-research.github.io/proteinfer/



