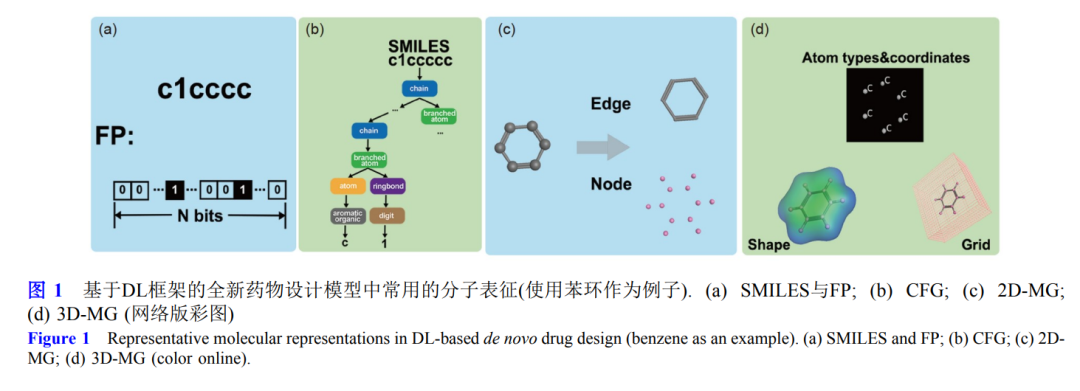
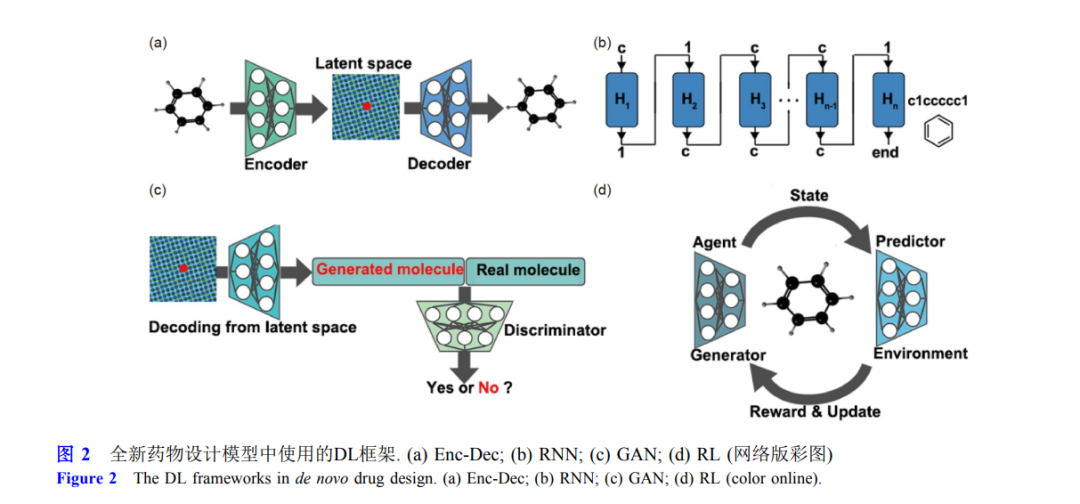
近年 来, 随着深度学习技术的不断发展, 基于深度学习的全新药物设计方法为先导化合物的发现带来新的契机, 前景巨大. 这些全新药物设计模型使用的深度学习框架包括编码-解码器、循环神经网络、生成对抗网络、强化学习 等. 本文综述了这些深度学习框架的基本原理、模型输入分子表征以及效果评测指标, 并对其在全新药物设计领 域的应用前景进行了展望.**引言 **新药研发是一项周期长、投入高、风险大的复杂 系统工程. 据美国药品研究与制造商协会报告显示[1], 自21世纪以来, 临床试验复杂性的提高以及药品市场 监管模式的变化, 新药研发的平均成本急剧增加, 2010~2015年间达到了平均26亿美元, 也对药物的多 属性优化及其对作用靶点机制的明确性提出了更高的 要求. 先导化合物的发现与优化在新药研发过程中至关 重要, 高质量的先导化合物能够大大缩短药物探索的 时间, 提高成药的可能性. 在先导化合物的设计过程 中, 要充分考虑候选分子的结构新颖性、生物活性、 靶标选择性、化学可合成性、成药性和安全性等, 这 些性质直接影响药物开发的成败, 因此先导化合物的 发现一直是创新药物研发的主要瓶颈. 传统的先导化 合物发现的方法主要基于对天然产物的结构修饰; 近 20年来, 随着计算机硬件、软件和算法的飞速发展, 高通量虚拟筛选和全新药物设计等计算机辅助药物设 计技术开始取代传统方法, 并大大减少了先导化合物 发现的时间和经济成本[2~4]. 虚拟筛选技术常用于从海 量化合物数据库中搜索潜在的候选分子; 因其主要针 对已注册过的分子(目前大部分化合物数据库为 107 ~109 量级[5,6]), 搜索的化学空间远小于现推测的类 药化学空间(1060量级[7]), 因此虚拟筛选技术并不能完 全发掘全新的化合物结构[8~10].全新药物设计(de novo drug design, 也被称为药物 从头设计)则使用分子生成方法对化合物结构不断优化, 使新生成化合物的预设属性获得最优解. 它不依赖 已有的化学数据库, 可实现对类药空间更加全面的探 索和发掘. 传统的全新药物设计方法通常与片段生长 法或遗传算法相结合[11~15], 但生成的分子往往难以同 时满足新颖性与多种理想属性(如生物活性、化学可 合成性、成药性等)的要求[16,17]. 深度学习(deep learning, DL)的引入为全新药物设计注入了新的活力. 作为 近期发展最快的人工智能技术, DL能够更高效地处理 数据, 对化合物属性深度特征的提取能力更强[18], 已 在不同的药物研发任务中取得了显著的成果[19~22]. 目 前已有越来越多的基于DL框架的全新药物设计模型 被开发出来, 旨在产生符合多种理想性质的新颖骨架 分子.基于DL的全新药物设计方法按照算法基本框架 主要分为四类, 包括编码-解码器(encoder-decoder, Enc-Dec)[23,24]、循环神经网络(recurrent neural network, RNN)[25~27]、生成对抗网络(generative adversarial network, GAN)[28]、强化学习(reinforcement learning, RL)[29,30]. 尽管近五年内已经开发出近百种基 于DL的全新药物设计模型, 但是这些模型均是在上述 四种DL框架下进行两个方面的改造: 分子表征以及多 目标优化策略. 基于DL框架的全新药物设计模型可以 使用不同的分子表征作为输入, 如SMILES (simplified molecule input line entry specification)[23,31]、分子指纹 (molecular fingerprint, MFP)[25]、二维(two dimension, 2D)[32,33]或三维(three dimension, 3D)[34,35]的分子图 (molecular graph, MG)等. 多目标优化则是使用不同 策略来同时优化生成分子的多种属性. 例如, 使用不同 的DL框架约束控制生成的步骤, 使得分子对某一靶标 保持活性的同时也具有很高的可合成性[30,36]. 目前已 经有研究者成功使用DL框架设计并合成出有活性的 苗头化合物[31,37,38], 且耗时远远小于传统的全新药物 设计方法, 潜力巨大. 本文将针对不同的DL算法在全 新药物设计方法开发中的应用进行回顾和总结, 以期 促进更多研究者的参与.