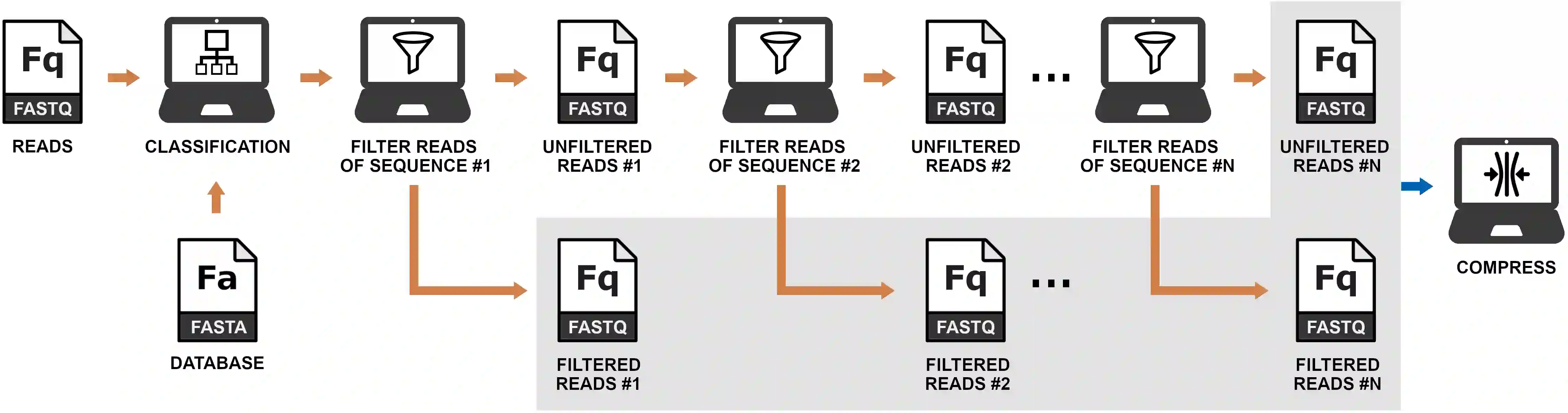
Minimizing data storage poses a significant challenge in large-scale metagenomic projects. In this paper, we present a new method for improving the encoding of FASTQ files generated by metagenomic sequencing. This method incorporates metagenomic classification followed by a recursive filter for clustering reads by DNA sequence similarity to improve the overall reference-free compression. In the results, we show an overall improvement in the compression of several datasets. As hypothesized, we show a progressive compression gain for higher coverage depth and number of identified species. Additionally, we provide an implementation that is freely available at https://github.com/cobilab/mizar and can be customized to work with other FASTQ compression tools.
翻译:暂无翻译