Nature子刊发表华人学者最新成果!整合高通量多组学数据,为复杂疾病遗传学研究打开新的大门
点击上方“公众号”可以订阅哦!

北京时间4月16日凌晨,国际权威学术期刊Nature Neuroscience发表了一篇题为“A Bayesian framework that integrates multi-omics data and gene networks predicts risk genes from schizophrenia GWAS data”的研究文章。在该文中,来自美国范德堡大学的研究人员建立了一个独特而巧妙的统计模型并利用此模型整合高通量多组学数据来剖析精神分裂症发病机理,将当前对精神分裂症的遗传学理解从DNA层面大大的拓深到了基因层面。其结果对于精神分裂症的发生机理和临床治疗有重要的指导意义。更重要的是,该文提出的统计模型是一个普适的方法,其应用可以推广到任何其他疾病,从而给人类复杂疾病的遗传学研究打开了一扇新的窗,其意义不言而喻。
此项研究由美国范德堡大学(Vanderbilt University)遗传中心的李冰山课题组完成,范德堡大学的汪泉博士和陈锐博士为文章的共同第一作者。

Nature Neuroscience为Nature旗下子刊,其影响因子为19.912,是国际顶级学术期刊,在Journal Citation Reports的261个神经科学类期刊中列第2位。
全基因组关联分析(Genome-wide association studies, GWAS)是目前人类复杂疾病研究中最常见的一种遗传学分析方法。其主要原理是通过比对患病病人和正常人基因组上的单核苷酸多态性(single nucleotide polymorphisms,SNP)来确定基因组中的哪些SNP位点与该疾病有统计学意义上的关联关系。虽然在很多疾病中,GWAS已被成功用于定位关联SNP,但是如何解释这些SNP的生物学意义一直是相关研究中的难点,主要有以下几点原因。
1. 由于实验设计的限制,GWAS中使用的SNP是通过计算挑选出来的最适合代表整个基因组的标签SNP(tag SNP)集合。绝大多数的情况下,它们并不是直接导致疾病的致病SNP(causal SNP)。
2. 在生物体中,控制生物性状基本遗传学单位是基因,行使生物功能的主要单位是基因的产物——蛋白质。通过GWAS得到的关联SNP绝大多数都位于基因组中的非编码区,这表示它们不能通过改变蛋白质的序列导致疾病,而是通过调控致病基因的表达行使它们的作用。如果只知晓关联SNP而不知道致病基因,就很难理解疾病的遗传学机理,从而严重阻碍GWAS成果的后续应用。
3. 生物体内染色体的存在形式并不是线性的,它们会进行各种高维的折叠和卷曲来实现非常丰富而复杂的调控关系,这也意味着关联SNP可以调控距离很远的基因。因此,如何确定关联SNP调控的致病基因并不是一件容易的事。
近年来,随着DNA测序技术的快速发展,高通量测序数据迅速积累,给这个问题的研究带来了新的机遇和挑战。如何整合并利用目前已经大量产生的多组学(基因组学、转录组学、蛋白质组学等)数据来帮助寻找致病基因是当前遗传学研究中的热点和难点,对理解人类复杂疾病的发病机理以及药物设计有重要的推进作用。
在该文中,研究人员利用数学工具,建立了一个独特而巧妙的贝叶斯模型,名为“iRIGS”(integrative RIsk Gene Selector),以整合高通量多组学数据和基因相互作用网络进行以基因为中心的统计推断。在模型的具体实现方面,他们创造性地引入了吉布斯抽样(Gibbs sampling)的概念来解决高维数据的计算难题(图1)。在当前的生物大数据时代,如何从浩瀚的数据库中挑选合适的组学数据来研究是个棘手的问题。在iRIGS中,研究人员提出了一个新颖的两步走的策略来解决这个难题。在第一步中,他们先用GWAS数据和基因相互作用网络预测出一组准致病基因,这组准致病基因会含有一些真正的致病基因,但是信噪比不高,有待提高。在第二步中,他们利用这组准致病基因作为准则来挑选合适的组学数据并整合进模型,从而达到预测更高可信度的致病基因的目的。
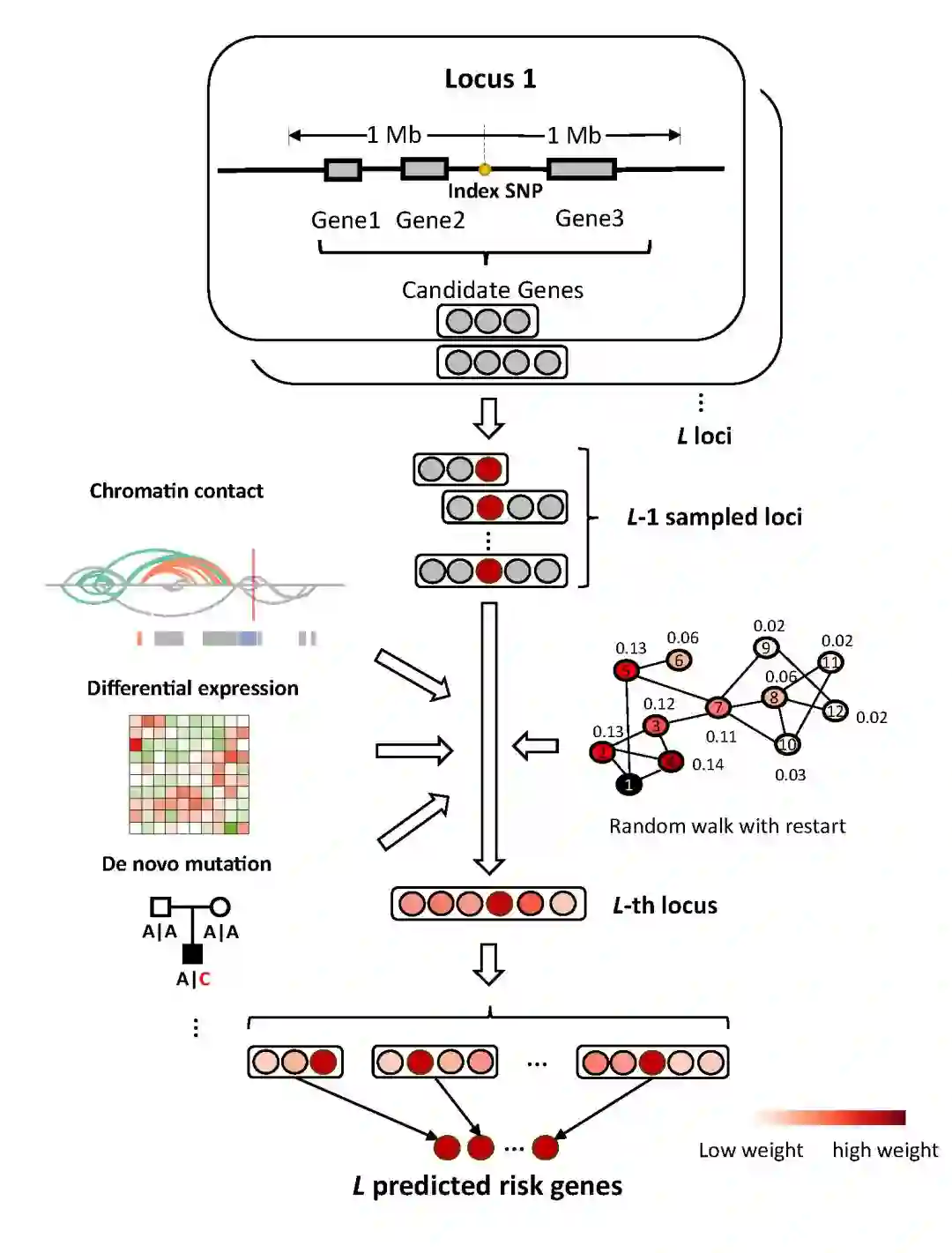
图1. iRIGS的基本框架
在精神分裂症的个案研究中,研究人员发现他们预测的准致病基因在新生突变(De novo mutation)中高度富集,在病人和对照组样本之间大量差异表达。同时,利用高通量染色体构象捕获(Hi-C)数据,研究人员发现准致病基因更容易与增强子(enhancer)等其他远程调控元件有相互作用(chromatin contact)。综合这些信息,研究人员在第二步中整合了基因组学(Genomics)、表观基因组学(Epigenomics)以及转录组学(Transcriptomics)等信息更精确的预测出了GWAS SNP调控的致病基因。相比于人体其他组织,这些基因在人脑相关的组织里更容易高表达并且深度参与大脑内部的神经信号传导过程。比如说,这些基因在钙离子通道基因中高度富集,而钙离子通道的紊乱是精神分裂症的一个重要致病诱因。从时间维度来看,相比于成人脑部组织,这些基因在胚胎发育过程中更活跃。另外,在基因敲除的小鼠模型实验中,研究人员发现被敲除这些基因的小鼠大多表现出跟中枢神经系统受损相关的表型。这些结果与目前认为精神分裂症是一种神经发展障碍性疾病(neurodevelopmental disorder)的主流认识高度一致。
从GWAS SNP推进到致病基因的重要性还体现于它给临床预防和治疗带来的巨大前景。由于复杂疾病的异质性(heterogeneity),不同病人患病的原因和机理可能不尽相同。如果在开发新的疾病预防和治疗方法时能将病人的个人致病基因,生活环境和生活习惯等因素都考虑在内的话,将会非常有助于提高疗效,这也是目前热门的国家战略项目——“精准医学计划”的主要目标。该研究中致病基因的精确定位为精准医疗提供了非常好的基础。这不仅能提高个人患病风险评估的准确性,帮助分析和诊断病人的患病机理,还对相关的药物设计有显著的推进作用。
以精神分裂症为例,目前针对该病的药物设计进展十分缓慢,其中一个重要的原因是对药物靶标基因的认知十分匮乏。因为药物研发是一项高投入高风险的产业,制药公司一直致力于发展新的策略来有效的提高产出投入比。已有研究表明,建立在遗传学分析基础上的药物研发工作有更高的成功率。此研究中得到的致病基因融合了来自遗传学和多基因组学方面的证据,是值得高度怀疑的候选靶标基因。除了研发新药,药物重定向(drug repositioning)也是目前药物设计领域的重要手段之一。药物重定向是基于已有药物的重新挖掘,因而能够有效地降低药物研发的周期、成本和风险,是未来突破新药研发高投入低产出困境的有效方法之一。利用全球最大的药物基因靶标数据库中的信息,研究人员发现这些致病基因在美国FDA认证的已知药物的靶标基因中高度富集,从而给针对精神分裂症的药物重定位带来了很大的希望。
还有一点值得强调的是,该文提出的模型和算法在数据分析方面十分的灵活,不仅能整合已有的高通量多组学数据,对未来新产生的多组学数据以及其他类型的生物学数据也具备强大的整合能力。这一点是目前该领域内其他常见的研究方法所做不到的。另外,该模型还是一个非常普适的方法,不仅仅可以用来研究精神分裂症,也可以用来研究任何其他复杂疾病。相信,随着未来多种多样的生物学数据的快速积累以及新的GWAS SNP的发现,iRIGS在各种复杂疾病的机理研究中会有越来越广泛的应用。
最后要强调的一点是,癌症作为复杂疾病的一种,有着更为复杂的机理。由于很多癌症具有很高的遗传性(heritability),使用相关的遗传学数据,该方法也可以推广到癌症的遗传学研究中。而同时肿瘤领域又有及其丰富的多组学数据,比如TCGA以及肿瘤药物开发相关的LINCS等大型资源。李冰山课题组和Thomas Jefferson University的杨虎山课题组在密切合作,依托于本模型来整合肿瘤的多组学数据,推动癌症的风险预测、早期诊断和药物筛查的发展。
注:投稿请电邮至124239956@qq.com ,合作 或 加入未来产业促进会请加:www13923462501 微信号或者扫描下面二维码:

文章版权归原作者所有。如涉及作品版权问题,请与我们联系,我们将删除内容或协商版权问题!联系QQ:124239956





