【哈佛大学最新论文】使用AlphaFold估算蛋白质模型精度的最新技术

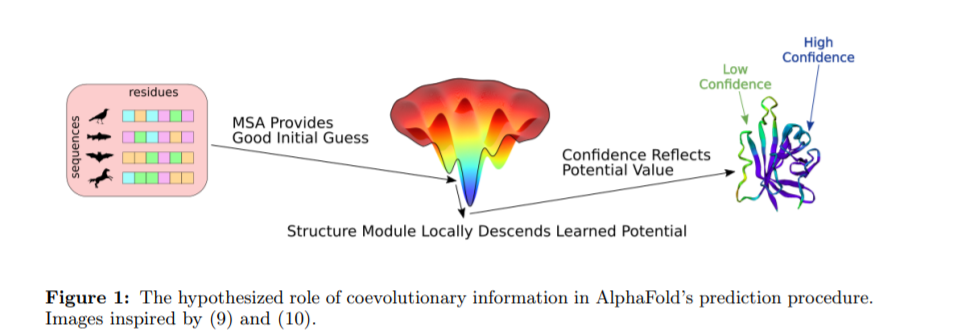
从蛋白质的初级氨基酸序列预测蛋白质的三维结构是结构生物学中一个长期存在的挑战。最近,AlphaFold等方法通过将深度学习技术与相关蛋白质序列多序列比对的共同进化数据相结合,在这项任务上取得了显著的性能。共同进化信息的使用对这些模型的准确性至关重要,没有这些信息,它们的预测性能会大幅下降。然而,在活细胞中,蛋白质的三维结构完全由其原始序列和生物物理定律决定,这些定律使蛋白质折叠成低能量的构型。因此,通过学习一种高度精确的生物物理能量函数,应该可以仅从其原始序列预测蛋白质的结构。我们提供证据表明AlphaFold已经学会了这样一个能量函数,并使用共同进化数据来解决寻找低能构象的全局搜索问题。我们证明,AlphaFold的学习电位功能可以用于以最先进的精确度对候选蛋白质结构的质量进行排序,而无需使用任何共同进化数据。最后,我们提出了一种方法,利用这个势函数来预测蛋白质结构,而不需要MSAs。
专知便捷查看
便捷下载,请关注专知公众号(点击上方蓝色专知关注)
后台回复“APMA” 就可以获取《【哈佛大学最新论文】使用AlphaFold估算蛋白质模型精度的最新技术》专知下载链接

请扫码加入专知人工智能群(长按二维码),或者加专知小助手微信(zhuanzhi02),加入专知主题群(请备注主题类型:AI、NLP、CV、 KG、论文等)交流~


登录查看更多
相关内容
专知会员服务
21+阅读 · 2022年3月14日
Arxiv
0+阅读 · 2022年4月19日
Arxiv
0+阅读 · 2022年4月18日


相关VIP内容
专知会员服务
21+阅读 · 2022年3月14日
相关资讯
相关论文
Arxiv
0+阅读 · 2022年4月19日
Arxiv
0+阅读 · 2022年4月18日